
Abstract
Acute kidney injury (AKI), characterized by aggressive inflammatory responses and destruction of renal resident cells, can cause abrupt kidney dysfunction. To date, effective therapy for AKI is lacking. In this study, we evaluated the renoprotective effect of wogonin, an herbal active compound, using a cisplatin-induced AKI mouse model. In vivo results show that wogonin substantially suppressed the increased levels of serum creatinine and blood urea nitrogen (BUN) almost to the normal level. Wogonin also attenuated tubular damage, shown by PAS staining, electron microscopy and molecular analysis of KIM-1. In addition, wogonin suppressed kidney inflammation as indicated by a >60% decrease in macrophage infiltration, a >50% reduction in inflammatory cytokine production and inhibited NF-κB activation in the injured kidney. Mechanistically, molecular docking results show that wogonin effectively inhibited RIPK1 by occupying the ATP-binding pocket of the enzyme, which is a key regulator of necroptosis. Moreover, inhibition of RIPK1, or RIPK3, reversed the protective effects of wogonin in cisplatin-treated HK2 cells, indicating wogonin works in a RIPK1/RIPK3-dependent manner. Surprisingly, wogonin enhanced the anti-proliferative effect of cisplatin on human hepatoma HepG2 cells. Thus, our findings suggest wogonin may be a renoprotective adjuvant for cisplatin-based anticancer therapy.
Similar content being viewed by others


Main
Acute kidney injury (AKI) is characterized by an abrupt decline in kidney function. Indeed, patients with AKI show high morbidity and mortality rates.1, 2, 3 Previous studies suggest AKI is triggered by renal ischemia reperfusion injury (IRI), nephrotoxic insult or sepsis.4, 5, 6 Pathological features of AKI include excessive inflammatory cell recruitment, over-production of inflammatory factors, death and delayed proliferation of renal resident cells.7, 8 However, detailed molecular mechanisms of AKI and effective therapies need further exploration.
Given positive pharmacological effects and low cytotoxicity, Traditional Chinese Medicine (TCM) has been used to treat many diseases in both laboratory and clinical settings. Here, we screened >10 TCM-derived active components, including aloin, barbaloin, icariin, protocatechuic acid, puerarin, sodium houttuyfonate, sophoridine, protocatechuic aldehyde, wogonin and wogonoside, for renoprotective effects against cisplatin-induced kidney injury (Supplementary Figure 1). We identified wogonin as the most potent compound, which significantly prevented cisplatin-induced injury on renal tubular epithelial cells by reducing cell death and inflammatory response.
Necroptosis is a newly identified cell death program recently reported as a key event in the pathogenesis of AKI. It has been detected in several types of AKI models.6, 9, 10 Previous studies show that genetically, or pharmacologically, blocking necroptosis pathway molecules, including receptor-interacting protein 1 (RIPK1), RIPK3 and mixed lineage kinase domain-like protein (MLKL), significantly inhibits necroptosis and attenuates renal injury. This suggests RIPK1/RIPK3/MLKL axis may be a potential therapeutic target for AKI.11, 12, 13 It is worth noting that necroptosis, not apoptosis, triggers a severe inflammatory response in AKI at the cellular level.14, 15
In this study, molecular docking experiments suggest wogonin inhibits the activity of RIPK1 enzyme by occupying the ATP-binding pocket. Thus, we hypothesized that wogonin protects against cisplatin-induced AKI by inhibiting necroptosis. As expected, we demonstrated that wogonin confers renoprotective effects on cisplatin-induced AKI via a RIPK1-dependent mechanism. Surprisingly, we found that wogonin further enhanced the antitumor activity of cisplatin on hepatoma HepG2 cells. This suggests wogonin serves as a renoprotective agent during cisplatin treatment on certain types of cancer patients.
Materials and methods
Murine Model of Cisplatin-Induced AKI
Mice were housed in the specific pathogen-free facility. The experimental protocols were approved by the Institutional Animal Experimentation Ethics Committee of Anhui Medical University. Animal model of cisplatin-induced AKI was established in male C57BL/6 mice (aged 6–8 weeks) by injection of cisplatin with a single dose at 20 mg/kg intraperitoneally, their littermates were injected with saline as normal control. Wogonin with concentrations of 12.5, 25 and 50 mg/kg was injected intraperitoneally daily, 3 days later, mice were killed under anesthesia, the kidney tissues and blood were collected 3 days later for further analysis. Blood was collected for detection of blood urea nitrogen (BUN) and creatinine according to the manufacturer’s instructions. Kidneys were harvested for paraffin embedding, molecular analysis and electron microscopy studies. Paraffin sections (4 mm) were stained with PAS or labeled with antibodies against TNF-α according to the manufacturer’s instructions (sc-374185, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Kidney damage was scored on PAS-stained kidney sections (n=6–8) by analyzing the percentage of tubules that displayed tubular necrosis, cast formation, and tubular dilation as follows: 0=normal; 1=10%; 2=10%–25%; 3=26%–50%; 4=51%–75%; 5=75%. Ten consecutive fields under power fields (× 20) was scored or quantified.
Reagent and Materials
Wogonin was purchased from Meilune Biology Technology (MB6663, CAS 632-85-9, DaLian, China). Necrostatin-1 (Nec-1) was obtained from Santa Cruz Biotechnology (sc-200142A). The antibodies specific for RIPK1, RIPK3, TNF-α, kidney injury molecular-1 (KIM-1) and β-actin were purchased from Santa Cruz Biotechnology (sc-7881, sc-135170, sc-374185, sc-53769, sc-130656). The antibody specific for F4/80 was purchased from Serotec (MCA497, Oxford, UK). According to the manufacturer’s instructions and our preliminary experiment, F4/80 antibody (1:100), KIM-1 antibody (1:200) and TNF-α antibody (1:200) were diluted in 1% BSA/PBS solution and then used in IHC and immunofluorescence staining respectively. Rabbit anti-p-MLKL, anti-cleaved-caspase-3 and anti-cleaved-caspase-8 were obtained from Cell Signaling Technology (14516 s, #9662, #4790, CST, Danvers, MA, USA). Lipofectamine 2000 was purchased from SciencBio Technology (Invitrogen, Carlsbad, CA, USA). Protein Assay Kit was bought from Beyotime Institute of Biotechnology (Jiangsu, China).
Cell Culture
Human tubular epithelial (HK2) cells were cultured in DMEM-F12 supplemented with 5% FBS, 37 °C in 5% CO2 atmosphere. After starving, cisplatin (20 μM) was added to cells for 24 h after pre-cultured with wogonin or NEC-1. Then, cells were harvested for further detection for cell viability, indexes of tubular injury (KIM-1), apoptosis (cleaved-caspase-3 and cleaved-caspase-8), necroptosis indexes (RIPK1, RIPK3 and p-MLKL) and inflammatory factors (TNF-α, IL-1β and IL-6) by western blot analysis and real-time PCR. Independent experiments were performed throughout the in vitro studies in triplicate or quadruplicate.
MTT Assay
Cell viability was detected by MTT (thiazole blue colorimetry) assay. The MTT assay is a colorimetric assay for assessing cell metabolic activity. NAD(P)H-dependent cellular oxidoreductase enzymes may, under defined conditions, reflect the number of viable cells present. These enzymes are capable of reducing the tetrazolium dye MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to its insoluble formazan, which has a purple color. Human HK2 cells were seeded in 96-well plates and pre-incubated with a set of concentrations of wogonin (arranged from 0.625 to 40 μg/ml) for 24 h, respectively, with or without administration of cisplatin for 24 h. Tumor cell lines, including hepatic cancer HepG2 and BEL-7402, and stomach cancer SGC-7901 cells, were seeded in 96-well plates and pre-incubated with wogonin (1.25 μg/ml) for 24 h with or without administration of cisplatin (20 μM, 24 and 48 h, respectively). Then, 5 mg/ml MTT solution was added to each well for 4 h at 37 °C. Supernatant was removed followed by adding 150 μl of DMSO to dissolve formazan crystals. The optical density (OD) was determined with microplate reader (Multiskan MK3, Thermo, USA) at 492 nm wavelength.
LDH Assay
The lactate dehydrogenase (LDH) cytotoxicity assay is a reliable colorimetric assay to quantitatively measure LDH released into the media from damaged cells as a biomarker for cellular cytotoxicity and cytolysis. Human HK2 cells were seeded in 96-well plates and pre-incubated with a set of concentrations of wogonin (arranged from 0.625 to 40 μg/ml) for 24 h, respectively, with or without administration of cisplatin for 24 h. Afterward, the release of LDH in cell culture supernant was detected by the manufacturer’s protocol (88953, Thermo Fisher Scientific, USA). LDH, a stable protein, exists in the cytoplasm of normal cells. Once the cell membrane is damaged, LDH is released to the extracellular spaces. LDH catalyzes the formation of pyruvate lactate, and reacted with INT (tetrazole salt) to form a purple crystalline material, then the OD was determined with microplate reader (Multiskan MK3, Thermo) at 500 nm wavelength.
Knockdown of RIPK3 in HK2 Cells by Transfecting shRNA Plasmid
RIPK3 shRNA (GeneChem Co. Ltd, Shanghai, China) was transfected into HK2 cells by using LipofectamineTM 2000 reagent (Invitrogen) according to the manufacturer’s protocol. A negative scrambled shRNA (GenePharma, Shanghai, China) was used accordingly. Briefly, equivoluminal shRNA and Lipofectamine 2000 were diluted in Opti-DMEM separately, and incubated for 5 min at room temperature. The diluted shRNA was combined within the diluted Lipofectamine 2000 and incubated for 15 min at room temperature in the dark. Finally, the mixture was applied to the HK2 cells. After incubation for 6 h, the cells were cultured with fresh DMEM-F12 supplemented with 5% FBS. Cells with RIPK3 shRNA screened by puromycin were cultured in incubator at 37 °C in 5% CO2 atmosphere.
RNA Extraction and Real-Time PCR
Total RNA was isolated from cultured HK2 cells or kidney tissues using the RNeasy Isolation Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Then real-time PCR was performed using the Bio-Rad iQ SYBR Green supermix with Opticon2 (Bio-Rad, Hercules, CA, USA) as previously described.16 The primers used in the current study were listed as follows:
Human IL-6, forward 5′-CGGGAACGAAAGAGAAGCTCTA-3′, reverse 5′- GAGCAGCCCCAGGGAGAA-3′; human TNF-α, forward 5′-CCCAGGGACCTCTCTCTAATCA-3′, reverse 5′- GCTACAGGCTTGTCACTCGG-3′; human KIM-1, forward 5′-CTGCAGGGAGCAATAAGGAG-3′, reverse 5′-TCCAAAGGCCATCTGAAGAC-3′; human β-actin, forward 5′-CGCCGCCAGCTCACCATG-3′, reverse 5′-CACGATGGAGGGGAAGACGG-3′; mouse IL-6, forward 5′-GAGGATACCACTCCCAACAGACC-3′, reverse 5′-AAGTGCATCATCGTTGTTCATACA-3′; mouse IL-1β, forward 5′-TGTGAAATGCCACCTTTTGA-3′, reverse 5′-GGTCAAAGGTTTGGAAGCAG-3′; mouse TNF-α, forward 5′- CATCTTCTCAAAATTCGAGTGACAA-3′, reverse 5′-TGGGAGTAGACAAGGTACAACCC-3′; mouse MCP-1, forward 5′- CTTCTGGGCCTGCTGTTCA-3′, reverse 5′-CCAGCCTACTCATTGGGATCA-3′; mouse KIM-1, forward 5′-CAGGGAAGCCGCAGAAAA-3′, reverse 5′-GAGACACGGAAGGCAACCAC-3′; mouse β-actin, forward 5′- CATTGCTGACAGGATGCAGAA-3′, reverse 5′-ATGGTGCTAGGAGCCAGAGC-3′.
PCR amplification was carried out over 40 cycles using the following conditions: denaturation at 95 °C for 20 s, annealing at 58 °C for 20 s and elongation at 72 °C for 20 s. The ratio for the mRNA of interest was normalized to β-actin and presented as the mean±s.e.m.
Western Blot Analysis
Cultured human HK2 cells and freshly isolated kidney tissue were lysed with RIPA lysate buffer, and the protein concentration was determined by using a BCA protein quantitative kit (Beyotime, Jiangsu, China). Then, western blot analysis was performed as described previously.16, 17 After blocking nonspecific binding with 5% BSA (room temperature, 1 h), membranes were consequently incubated with the primary antibody against KIM-1, RIPK1, RIPK3, p-MLKL, cleaved-caspase-3, cleaved-caspase-8 and β-actin overnight at 4 °C, followed by treated with IRDye 800-conjugated secondary antibody (Rockland immunochemicals, Gilbertsville, PA, USA). Signals were detected with LiCor/Odyssey infrared image system (LI-COR Biosciences, Lincoln, NE, USA) and the intensities of bands were quantified by using the Image J software (NIH, Bethesda, MD, USA).
Immunofluorescence
HK2 cells were cultured in eight-chamber glass slides in the presence or absence of cisplatin (20 μM) for 24 h after incubation with wogonin overnight. Cells were then fixed in 2% paraformaldehyde and incubated with the antibodies detecting KIM-1 or TNF-α, respectively (4 °C overnight), followed by 1 h of incubation with goat anti-rabbit IgG-rhodamine (Bioss Biotechnology, Bei Jing, China). Cells were then counterstained with DAPI and analyzed under fluorescence microscope (Zeissspot; Carl Zeiss Micro Imaging GmbH, Gottingen, Germany). The positive cells were counted from 500 nucleated cells and expressed as percentage of positive cells scored.
Transmission Electron Microscopy
To observe and evaluate the ultrastructural change in vivo and in vitro, kidney tissue or cultured HK2 cells was fixed in 2.5% glutaraldehyde and 1% osmic acid, stained with 1% uranylacetate and embedded in epoxy resin (EPON). Specimens were then detected by a transmission electron microscope (H-7700, Tokyo, Japan).
The Molecular Docking Analysis
The X-ray crystal structure of RIPK1 was obtained from the RCSB Protein Data Bank (PDB code: 4NEU). The ligand was constructed in SYBYL 7.3.5 (Tripos Inc., St Louis, MO, USA) and was charged using the Gasteiger–Huckel computational method. The docking studies were predicted using AUTODOCK 4.1 software (The Scripps Research Institute, La Jolla, CA, USA). The dimensions of the active site box were selected to be large enough to contain the entire protein molecule. All docking calculations were performed using a Lamarckian genetic algorithm. The most likely conformation of the ligand was selected based on its binding affinity and optimal binding arrangement with the hydrophobic centers and H-bond acceptors of RIPK1.
Statistical Analyses
Data are expressed as the mean±s.e.m. and analyzed using one-way analysis of variance (ANOVA), followed by Tukey post-hoc tests using GraphPad Prism 5.
Results
Wogonin Prevented Cisplatin-Induced Cell Death in Renal Tubular Epithelial Cells
MTT assay results show that wogonin had no inhibitory effect on human renal tubular epithelial (HK2) cells at a concentration <2.5 μg/ml (Figure 1a). Thus, we used concentrations of 0.625, 1.25 and 2.5 μg/ml for this study. MTT assay results show that cisplatin (20 μM) significantly inhibited cell viability of HK2 cells (>50%), which was restored by co-treatment with wogonin (Figure 1b). In contrast, LDH assay results show that cisplatin-induced cell death, and addition of wogonin reduced the number of injured cells in a dose-dependent manner (Figure 1c). As we all know, cisplatin is commonly used as an antitumor drug in clinical. We also tested whether wogonin would limit the antitumor activity of cisplatin in three solid tumor cell lines, including HepG2, BEL-7402 and SGC-7901. These solid tumor cell lines were seeded in 96-well plates and pre-incubated with wogonin (1.25 μg/ml) for 24 h with or without administration of cisplatin (20 μM, 24 and 48 h respectively). MTT assay results show that treatment of wogonin reduced the number of tumor cells in all tumor cell lines, suggesting wogonin may possess antitumor functions (Figure 1d). Furthermore, we found wogonin promoted the tumor cell killing effect of cisplatin (20 μM) on HepG2 cells, a commonly used hepatoma tumor cell line, at 48 h, but had no impact on hepatic cancer BEL-7402 or stomach cancer SGC-7901 cell lines. This suggests that the impact of wogonin on the antitumor effect of cisplatin is related to cell type and time of exposure.

Effect of wogonin on cell viability of cisplatin-treated HK2 and tumor cells. (a) Effect of different concentrations of wogonin on viability of HK2 cells by MTT assay. (b) Wogonin restored cell viability in cisplatin-treated HK2 cells (MTT assay). (c) Wogonin attenuated cisplatin-induced cytotoxicity (LDH) assay. (d) Wogonin did not attenuate the killing effects of cisplatin in three solid tumor cells lines including HepG2, BEL-7402 and SGC-7901 (MTT assay). Independent experiments were performed throughout the in vitro studies in triplicate or quadruplicate. *P<0.05, ***P<0.001 compared with the control. #P<0.05, ##P<0.01, ###P<0.001 compared with cisplatin-treated group. Cis, cisplatin; Wog, wogonin.
Wogonin Inhibited Cisplatin-Induced Necroptosis and Apoptosis in HK2 Cells
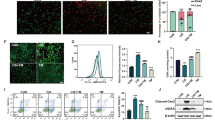
Representative electron microscope images show wogonin (1.25 μg/ml) inhibited nuclear swelling and loss of cell organelle content in cisplatin-treated HK2 cells (Figure 2a and Supplementary Figure 2). Flow cytometry and immunofluorescence of PI/AnnexinV results show that cisplatin-induced both apoptosis and necrosis in HK2. Data of flow cytometry and immunofluorescence consistently indicated that no <20% tubular epithelial cells suffer programmed cell death in response to cisplatin (20 μM). However, wogonin treatment reduced the percentages of both AnnexinV-FITC+/PI+ (late apoptotic or necrotic) cells and AnnexinV-FITC+/PI- (early apoptotic) cells (Figures 2b and c).

Wogonin reduces cisplatin-induced programmed cell death in HK2 cells. (a) Electron microscope. Results indicated that wogonin attenuated nuclear swelling and loss of cell organelle content in cisplatin-stimulated HK2 cells; (b, c) Flow cytometry and Immunofluorescence of PI/AnnexinV. Results showed that wogonin reduced the percentage of both cisplatin-induced apoptotic and necrotic HK2 cells. ***P<0.001 compared with the control. ##P<0.01 compared with cisplatin-treated group. Cis, cisplatin; Wog, wogonin.
Wogonin Suppressed Cisplatin-Induced HK2 Cell Damage and Inflammatory Response
We analyzed KIM-1 to evaluate whether wogonin effectively attenuated renal injury at the molecular level. Results from western blot analysis and quantitative data show that cisplatin-induced the expression of KIM-1, but wogonin (1.25 μg/ml) suppressed KIM-1 nearly back to baseline levels (Figures 3c and d). The suppressive effect of wogonin on KIM-1 was further confirmed by immunofluorescence and semi-quantification of KIM-1 in cisplatin-treated HK2 cells (Figures 3a and b). We also measured the effect of wogonin on cisplatin-induced inflammatory response in HK2 cells. Real-time PCR results show cisplatin significantly induced mRNA levels of TNF-α and IL-1β.13 However, wogonin (1.25 μg/ml) significantly suppressed both (Figure 3g). These findings were further confirmed by immunofluorescence and quantitative analysis of TNF-α at the protein level (Figures 3e and f). Finally, ELISA results show that pre-incubation with wogonin reduced protein levels of IL-6, IL-8 and MCP-1 in cisplatin-treated HK2 cells (Figure 3h).

Wogonin reduces cisplatin-induced KIM-1 level and inflammatory response in HK2 cells. (a, b) Immunofluorescence and quantification of KIM-1 in HK2 cells. Results indicated that treatment of wogonin significantly reduced protein level of KIM-1 in cisplatin-treated HK2. (c, d) Western blot analysis and quantitative data of KIM-1 in HK2 cells. (e, f) Immunofluorescence and quantification of TNF-α in HK2 cells. Results showed that treatment of wogonin decreased percentage of TNF-α-positive cells in response to cisplatin. (g) Real-time PCR in HK2 cells. Results showed that treatment of wogonin largely reduced cisplatin-induced mRNA levels of TNF-α and IL-1β. (h) ELISA in HK2 cells. Results indicated that treatment of wogonin significantly reduced cisplatin-upregulated protein levels of IL-6, IL-8 and MCP-1in HK2 cells. Independent experiments were performed throughout the in vitro studies in triplicate or quadruplicate. *P<0.05, **P<0.01, ***P<0.001 compared with the control. #P<0.05, ##P<0.01, ###P<0.001 compared with cisplatin-treated group. Cis, cisplatin; Wog, wogonin.
Wogonin Attenuated RIPK1 Signaling in Cisplatin-Treated HK2 Cells
We used western blot analysis and quantitative data to show pre-incubation of wogonin (1.25 μg/ml) reduced the protein levels of cleaved-caspase-3 and cleaved-caspase-8 by 18% and 16%, respectively (Figures 4a and b). Molecular docking is a progress of rigid docking. The conformation of macromolecular receptor is fixed, and small molecule ligand will have no impact on the protein conformation. The combination between small molecule ligand and amino-acid residues at active pocket is the role of molecular hydrogen key, hydrophobic key and so on. Figure 4d(B) is the amplification of the ligand-binding active pocket region of Figure 4d(A) in order to better observe the interaction of wogonin with amino-acid residues of RIPK1. Molecular docking data show that wogonin sits deeply in the allosteric pocket, formed by the DLG-out conformation and partially defined by the C-helix. The flavone structure is surrounded by hydrophobic amino acids, which interact through van der Waals contacts. The pyran ring is formed by one hydrogen bond between a carbonyl and the backbone amide of Glu63 on the c-helix. 5-Hydroxyl in the flavone structure also forms a hydrogen bond with Glu63. 7- hydroxyl in the flavone structure is formed by the third hydrogen bond with the amino acid Asp156 on the DLG motif. In the active state of kinase RIPK1, Glu63 in the alpha C-helix forms a salt bridge with Lys45, stabilizing ATP through H-bonds with the α- and β-phosphates. Importantly, this conserved DLG (Asp-Leu-Gly) was partially obstructed in the ATP-binding site. Our docking results show that wogonin occupies the ATP-binding pocket of the enzyme, and inhibits RIPK1 activity (Figure 4d). Western blot data suggest wogonin suppressed cisplatin-induced RIPK1, leading to the reduction of RIPK3 and phospho-MLKL in HK2 cells (Figure 4c).

Wogonin gently reduces caspase signaling while significantly suppressing RIP signaling in cisplatin-treated HK2 cells. (a) Western blot analysis and quantitative data of cleaved caspase-3 in cisplatin-treated HK2 cells. (b) Western blot analysis and quantitative data of cleaved caspase-8 in cisplatin-treated HK2 cells. Results of western blot demonstrated that treatment of wogonin gently decreased cisplatin-induced cleavage of caspase-3 and caspase-8 in HK2 cells. (c) Western blot analysis and quantitative data of RIPK1, RIPK3, p-MLKL in cisplatin-treated HK2 cells. Results showed that administration of wogonin substantially blocked RIPK1/RIPK3/MLKL in cisplatin-stimulated HK2 cells. (d) Molecular docking. Results of molecular docking indicated that wogonin may inhibit RIPK1 activity by occupying its ATP-binding pocket. Independent experiments were performed throughout the in vitro studies in triplicate or quadruplicate. *P<0.05, **P<0.01 compared with the control. #P<0.05, ##P<0.01, ###P<0.001 compared with cisplatin-treated group. Cis, cisplatin; Wog, wogonin.
Wogonin Protected Against Cisplatin-Induced Injury via a RIPK1-Dependent Mechanism
To further evaluate the role of RIPK1/RIPK3 in the inhibitory effect of wogonin in cisplatin-induced renal injury, we blocked RIPK1 and RIPK3. We found after NEC-1 (25μM) treatment, a RIPK1 inhibitor, RIPK1 levels decreased in the wogonin (1.25 μg/ml) treated group (Figure 5a). However, when RIPK1 was inhibited, wogonin failed to further suppress KIM-1 or IL-6. This indicates wogonin functions selectively through RIPK1 (Figures 5b and c). We also silenced the downstream pathway by transfecting a RIPK3 shRNA plasmid (Figure 6a). RIPK3 knockdown reduced RIPK3 and p-MLKL protein levels without affecting RIPK1 (Figure 6b). In addition, we found that RIPK3 inhibition significantly decreased the levels of KIM-1, TNF-α and IL-6 that were induced by cisplatin, but wogonin did not further down-regulate them in the absence of RIPK3 (Figures 6c and d). Taken together, these results suggest that wogonin functions via RIPK1/RIPK3-dependent mechanisms.

Wogonin fails to further reduce cisplatin-induced cell injury and inflammatory response in HK2 cells where RIPK1 is blocked. (a) Western blot analysis and quantitative data of RIPK1/RIPK3/MLKL signaling in Nec-1-treated HK2 cells. (b) Western blot analysis and quantitative data of KIM-1. Results indicated that when RIPK1 was blocked, wogonin failed to further suppress the protein levels of KIM-1. (c) Real-time PCR showed that when RIPK1 was blocked, wogonin failed to further decrease IL-6 in mRNA level. Independent experiments were performed throughout the in vitro studies in triplicate or quadruplicate. *P<0.05, **P<0.01, ***P<0.001 compared with the control. ##P<0.01, ###P<0.001 compared with cisplatin-treated group. $$P<0.01, $$$P<0.001 compared with Nec-1 non-treatment group. Cis, cisplatin; Wog, wogonin.

Wogonin fails to further reduce cisplatin-induced cell injury and inflammatory response in HK2 cells where RIPK3 is disrupted. (a) Western blot analysis and quantitative data of RIPK3 in HK2 cells. Results showed that RIPK3 was downregulated by transfection of RIPK3 shRNA plasmid. (b) Western blot analysis and quantitative data of RIPK1/RIPK3/MLKL signaling in RIPK3 knockdown HK2 cells. (c) Western blot analysis and quantitative data of KIM-1. Results indicated that when RIPK3 was knockdown, wogonin failed to further decrease the protein levels of KIM-1. (d) Real-time PCR identified that when RIPK3 was disrupted, wogonin failed to further decrease mRNA levels of TNF-α and IL-6. Independent experiments were performed throughout the in vitro studies in triplicate or quadruplicate. **P<0.01,***P<0.001 compared with the control. #P<0.05, ##P<0.01, ###P<0.001 compared with cisplatin-treated group. $$P<0.01,$$$P<0.001 compared with RIPK3 EV group. Cis, cisplatin; Wog, wogonin; EV, empty vector; KD, knockdown.
Wogonin Protected Against Cisplatin-Induced AKI
We further evaluated the in vivo renoprotective effect of wogonin on AKI. Our results from PAS staining show wogonin, in concentrations of 12.5, 25 and 50 mg/kg/day, significantly reduced tubular necrosis, tubular dilation and cast formation (Figure 7a). The therapeutic effects of wogonin in vivo were further confirmed by detection of serum creatinine and BUN (Figures 7b and c). Moreover, western blot analysis and real-time PCR also consistently show that wogonin downregulated cisplatin-induced KIM-1 protein and mRNA levels (Figure 7d). Given that wogonin (12.5 mg/kg/day) sufficiently blocked renal injury, this concentration was used to analyze the mechanisms by which wogonin protects against AKI in vivo.

Wogonin prevents cisplatin-induced renal injury and decline of renal function in vivo. (a) PAS staining and score. Results of PAS staining and score of severity indicated that treatment with a set of concentrations of wogonin alleviated tubular necrosis, tubular dilation and cast formation in cisplatin nephropathy. (b) Serum creatinine. (c) BUN. Results of serum creatinine and BUN showed that treatment of wogonin restored renal function in cisplatin nephropathy. (d) Western blot analysis and real-time PCR of KIM-1. Results indicated that treatment of wogonin in a set of concentrations substantially inhibit cisplatin-induced KIM-1 in both protein and mRNA levels. Data represent the mean±s.e.m. for 6–8 mice. **P<0.01, ***P<0.001 compared with control. ##P<0.01, ###P<0.001 compared with model. Cis, cisplatin; Wog, wogonin.
Wogonin Inhibited the Inflammatory Response and Tubular Cell Death in AKI by Targeting RIPK1-Mediated Necroptosis
Our immunohistochemistry data of F4/80 show that wogonin (12.5 mg/kg/day) decreased the number of infiltrated macrophages (Figure 8a). We also found TNF-α mRNA and protein levels were suppressed, and significantly reduced mRNA levels of IL-1β, IL-6 and MCP-1 (Figures 8b and c). Results from western blot analysis show wogonin inhibited phosphorylation of P65 NF-κB in cisplatin-injured kidneys (Figure 8d). Images from electron microscopy show destroyed nuclear structures and abnormal cell organelle content in necrotic tubular epithelial cells, and wogonin attenuated these damages (Figure 9a). Finally, we clarified the underlying mechanism by measuring RIPK1 signaling. Western blot analysis and quantitative data showed that wogonin substantially suppressed protein levels of RIPK1, RIPK3 and phospho-MLKL in cisplatin-injured kidneys (Figure 9b).

Wogonin attenuates cisplatin-induced renal inflammation in vivo. Immunohistochemistry of f4/80+ macrophages. IHC result and quantitative data indicated that treatment of wogonin prevented the infiltration of macrophages in injured kidney. (b) Immunohistochemistry of TNF-α. IHC result and quantitative data indicated that treatment of wogonin reduced the percentage of TNF-α+ cells in injured kidney. (c) Real-time PCR of inflammation indexes. Real-time PCR demonstrated that treatment of wogonin largely blocked upregulated mRNA levels of TNF-α, IL-1β, IL-6 and MCP-1 in cisplatin-injured kidney. (d) Western blot analysis showed that wogonin suppressed phospho-P65 level in the cisplatin-injured kidney (Figure 8d). Data represent the mean±s.e.m. for 6–8 mice. **P<0.01, ***P<0.001 compared with control. ##P<0.01, ###P<0.001 compared with model. Cis, cisplatin; Wog, wogonin.

Wogonin prevents RIPK1-mediated necroptosis in cisplatin nephropathy. (a) Electron microscope. Results clearly demonstrated that wogonin largely reduced nuclear structure damage and abnormal cell organelle content in injured kidney. (b) Western blot analysis of RIPK1/RIPK3/MLKL signaling. Results showed that treatment of wogonin significantly suppressed the activation of RIPK1/RIPK3/MLKL axis in cisplatin-injured kidney. Data represent the mean±s.e.m. for 6–8 mice. ***P<0.001 compared with control. ##P<0.01, ###P<0.001 compared with model. Cis, cisplatin; Wog, wogonin.
Discussion
In the present work, we demonstrated the renoprotective effects of wogonin, an active component of TCM, on cisplatin-induced injury by targeting RIPK1-mediated necroptosis in vivo and in vitro. AKI is associated with high mortality, especially in hospitalized patients.1, 2 AKI also leads to chronic diseases under certain conditions.18, 19, 20 To date, effective therapies for AKI are lacking. Given that AKI is characterized and mediated by aggravated inflammation and excessive cell death, we presumed that therapeutic reagents targeting inflammation and/or cell death-related signaling or molecules would suppress AKI. Therefore, we evaluated a number of TCM monomers, which have shown anti-inflammatory properties in other organs and in vitro systems. Preliminary studies suggest wogonin, a major component of Scutellaria baicalensis Georgi, has potent renoprotective effects on cisplatin-induced cell death and inflammatory response in HK2 cells compared with other TCM monomers. We consequently verified its therapeutic effects and working mechanism by using a cisplatin-induced AKI mouse model. Previous studies showed wogonin has pharmacological effects on inflammation,21, 22, 23 apoptosis,24 oxidative stress25, 26 and cell cycle regulation,27 but the related mechanisms have not been fully elucidated. Here, we found that wogonin significantly suppressed RIPK1 to attenuate necroptosis.
Necroptosis, the best-characterized regulated necrosis, is a recently identified cell death program that has an important role in AKI.6, 28 Cells that undergo necroptosis not only show necrosis-like structural characteristics, but are also regulated by the death signaling pathway.8, 12 Necroptosis can be initiated by various stimuli, including TNF, FASL, TRAIL, anticancer drugs and Toll-like receptors (TLRs).29, 30, 31 Typically, TNF-induced cell death is mediated by the three main complexes: complex IIa (traddosome), IIb (ripoptosome) and IIc (necrosome).30 Complex IIa consists of TRADD, FADD and caspase-8 and functions in initiating apoptosis. Complex IIb contains RIPK1, FADD, FLIPL and caspase-8 and promotes RIPK1-dependent apoptosis. The major components for complex IIc include FADD, FLIPL, RIPK1 and RIPK3, as well as inactivated caspase-8. Complex IIc is responsible for RIPK1 and RIPK3-dependent necroptosis.32, 33 RIPK3 activation induced the phosphorylation, oligomerization and translocation of MLKL to membranes, thereby leading to permeabilization of plasma membrane and cell death. It is of note that RIPK3 is a well-known and major upstream molecule of MLKL, but the activation mechanism and function of MLKL are of interest and need to be further investigated.34, 35 Until now, RIPK1/RIPK3/MLKL axis has been well accepted as the predominant mediator in necroptosis.11, 36
In kidney cells, both apoptosis and necroptosis occur during the progression of AKI, especially in proximal tubules.37, 38, 39 Importantly, emerging evidence indicates that administration of Nec-1, but not Z-VAD, prevents renal IRI.9, 40 Nec-1 also attenuates renal tubular injury in cisplatin or contrast-induced AKI and kidney injury post-transplantation.41, 42 This was further confirmed by in vitro findings showing that treatment of Nec-1 attenuated cisplatin, cyclosporin A or ischemia-induced death of tubular epithelial cells.41, 43, 44, 45 In line with these observations, knockout of downstream RIPK3 or MLKL also prevented kidney injury in ischemic and cisplatin-induced AKI models.13, 39 Taken together, these findings indicate the essential role of RIP-mediated necroptosis in pathogenesis of various AKIs. In the current study, we identified wogonin as an ideal inhibitor for RIPK1 given it binds to the ATP-binding pocket of the enzyme to inhibit RIPK1 activity. Treatment of wogonin largely inhibited necroptosis while gently attenuating apoptosis of renal tubular epithelial cells, and consequently prevented renal damage and decline of renal function effectively in a cisplatin-induced AKI model.
In addition, our results show that wogonin confers anti-inflammatory effects on cisplatin-triggered renal inflammation. Wogonin significantly suppressed the release of pro-inflammatory cytokines including TNF-α, IL-1β and IL-6. Indeed, necroptosis is much more important to mediate inflammatory response compared with apoptosis given destruction of cell membranes leads to release of endogenous pro-inflammatory molecules. This process is known as damage-associated molecular patterns (DAMPs), which include high-mobility group box 1, heat-shock proteins, uric acid, IL-33.46 These DAMPs activate innate immunity and promote the release of pro-inflammatory cytokines by interacting with receptors like TLRs and initiate downstream inflammation-related signaling.47, 48 The enhanced renal inflammation, necroinflammation, leads to more necrosis in the injured kidney, forming a positive-feedback loop.12, 14, 15 In this regard, prevention of both necroptosis and inflammation appears beneficial for AKI treatment. Interestingly, RIPK1 also has a role in the inflammatory response by activating NF-κB, which is a central regulator in renal inflammation and cellular context-dependent mechanisms.49, 50 Recent studies, and our results, consistently show that wogonin suppresses NF-κB signaling,51, 52 which may be mediated by inhibition of RIPK1.
In the present study, we also found that wogonin did not attenuate the antitumor effect of cisplatin. Thus, it may promote cisplatin-induced tumor cell death in certain types of tumor cells. This finding is in line with recent studies that showed wogonin sensitized A549 cells, HeLa cells, and two cisplatin-resistant head and neck cancer (HNC)cell lines (AMC-HN4R and -HN9R) in response to cisplatin. Those data show that wogonin potentiated cisplatin-induced cancer cell apoptosis by accumulating intracellular reactive oxygen species.53 Further, targeting Nrf2 with wogonin overcomes cisplatin resistance in HNC,54 indicating wogonin is an ideal renoprotective agent for cancer patients undergoing cisplatin treatment.
Collectively, our present study identified renoprotective effects of wogonin on cisplatin-induced AKI by targeting RIPK1-dependent necroptosis. Wogonin preserved, and even promoted, the tumor-killing effect of cisplatin in different types of tumor cells. Thus, wogonin should be further evaluated as a renoprotective adjuvant for AKI in cisplatin-treated cancer patients.
References
Thomas ME, Blaine C, Dawnay A et al. The definition of acute kidney injury and its use in practice. Kidney Int 2015;87:62–73.
Waikar SS, Liu KD, Chertow GM . Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol 2008;3:844–861.
Yang L, Xing G, Wang L et al. Acute kidney injury in China: a cross-sectional survey. Lancet 2015;386:1465–1471.
Chertow GM, Burdick E, Honour M et al. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 2005;16:3365–3370.
Allison SJ . Acute kidney injury: aiming to enhance debris clearance and improve outcomes in AKI. Nat Rev Nephrol 2016;12:123.
Linkermann A . Nonapoptotic cell death in acute kidney injury and transplantation. Kidney Int 2016;89:46–57.
Zuk A, Bonventre JV . Acute kidney injury. Annu Rev Med 2016;67:293–307.
Sancho-Martinez SM, Lopez-Novoa JM, Lopez-Hernandez FJ . Pathophysiological role of different tubular epithelial cell death modes in acute kidney injury. Clin Kidney J 2015;8:548–559.
Linkermann A, Brasen JH, Himmerkus N et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int 2012;81:751–761.
Gao L, Wu WF, Dong L et al. Protocatechuic aldehyde attenuates cisplatin-induced acute kidney injury by suppressing Nox-mediated oxidative stress and renal inflammation. Front Pharmacol 2016;7:479.
Linkermann A, Chen G, Dong G et al. Regulated cell death in AKI. J Am Soc Nephrol 2014;25:2689–2701.
Wang S, Zhang C, Hu L et al. Necroptosis in acute kidney injury: a shedding light. Cell Death Dis 2016;7:e2125.
Xu Y, Ma H, Shao J et al. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol 2015;26:2647–2658.
Linkermann A, Stockwell BR, Krautwald S et al. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol 2014;14:759–767.
Mulay SR, Linkermann A, Anders HJ . Necroinflammation in kidney disease. J Am Soc Nephrol 2016;27:27–39.
Meng XM, Huang XR, Chung AC et al. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol 2010;21:1477–1487.
Meng XM, Huang XR, Xiao J et al. Diverse roles of TGF-beta receptor II in renal fibrosis and inflammation in vivo and in vitro. J Pathol 2012;227:175–188.
Bucaloiu ID, Kirchner HL, Norfolk ER et al. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int 2012;81:477–485.
Goldstein SL, Jaber BL, Faubel S et al. AKI transition of care: a potential opportunity to detect and prevent CKD. Clin J Am Soc Nephrol 2013;8:476–483.
Rewa O, Bagshaw SM . Acute kidney injury-epidemiology, outcomes and economics. Nat Rev Nephrol 2014;10:193–207.
Chen P, Zhou X, Zhang L et al. Anti-inflammatory effects of Huangqin tang extract in mice on ulcerative colitis. J Ethnopharmacol 2015;162:207–214.
Mabalirajan U, Ahmad T, Rehman R et al. Baicalein reduces airway injury in allergen and IL-13 induced airway inflammation. PLoS ONE 2013;8:e62916.
Zhang L, Ren Y, Yang C et al. Wogonoside ameliorates lipopolysaccharide-induced acute lung injury in mice. Inflammation 2014;37:2006–2012.
Zhang L, Wang H, Cong Z et al. Wogonoside induces autophagy-related apoptosis in human glioblastoma cells. Oncol Rep 2014;32:1179–1187.
Qian C, Wang Y, Zhong Y et al. Wogonin-enhanced reactive oxygen species-induced apoptosis and potentiated cytotoxic effects of chemotherapeutic agents by suppression Nrf2-mediated signaling in HepG2 cells. Free Radic Res 2014;48:607–621.
Wozniak D, Drys A, Matkowski A . Antiradical and antioxidant activity of flavones from Scutellariae baicalensis radix. Nat Prod Res 2015;29:1567–1570.
Kim D, Lee AS, Jung YJ et al. Tamoxifen ameliorates renal tubulointerstitial fibrosis by modulation of estrogen receptor alpha-mediated transforming growth factor-beta1/Smad signaling pathway. Nephrol Dial Transplant 2014;29:2043–2053.
Linkermann A, De Zen F, Weinberg J et al. Programmed necrosis in acute kidney injury. Nephrol Dial Transplant 2012;27:3412–3419.
Jouan-Lanhouet S, Riquet F, Duprez L et al. Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol 2014;35:2–13.
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S et al. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014;15:135–147.
Vanlangenakker N, Vanden Berghe T, Vandenabeele P . Many stimuli pull the necrotic trigger, an overview. Cell Death Differ 2012;19:75–86.
Cho YS, Challa S, Moquin D et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009;137:1112–1123.
Zhang DW, Shao J, Lin J et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009;325:332–336.
Sun L, Wang H, Wang Z et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012;148:213–227.
Petrie EJ, Hildebrand JM, Murphy JM . Insane in the membrane: a structural perspective of MLKL function in necroptosis. Immunol Cell Biol 2017;95:152–159.
Tait SW, Oberst A, Quarato G et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep 2013;5:878–885.
Linkermann A, Himmerkus N, Rolver L et al. Renal tubular Fas ligand mediates fratricide in cisplatin-induced acute kidney failure. Kidney Int 2011;79:169–178.
Safirstein RL . Am I my brother's keeper?: fratricide in the kidney. Kidney Int 2011;79:149–150.
Linkermann A, Brasen JH, Darding M et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA 2013;110:12024–12029.
Price PM, Hodeify R . A possible mechanism of renal cell death after ischemia/reperfusion. Kidney Int 2012;81:720–721.
Tristao VR, Goncalves PF, Dalboni MA et al. Nec-1 protects against nonapoptotic cell death in cisplatin-induced kidney injury. Ren Fail 2012;34:373–377.
Linkermann A, Heller JO, Prokai A et al. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J Am Soc Nephrol 2013;24:1545–1557.
Ouyang Z, Zhu S, Jin J et al. Necroptosis contributes to the cyclosporin A-induced cytotoxicity in NRK-52E cells. Pharmazie 2012;67:725–732.
Zhang L, Jiang F, Chen Y et al. Necrostatin-1 attenuates ischemia injury induced cell death in rat tubular cell line NRK-52E through decreased Drp1 expression. Int J Mol Sci 2013;14:24742–24754.
Tristao VR, Pessoa EA, Nakamichi R et al. Synergistic effect of apoptosis and necroptosis inhibitors in cisplatin-induced nephrotoxicity. Apoptosis 2016;21:51–59.
Scaffidi P, Misteli T, Bianchi ME . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002;418:191–195.
Vilaysane A, Chun J, Seamone ME et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol 2010;21:1732–1744.
He S, Liang Y, Shao F et al. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA 2011;108:20054–20059.
Kelliher MA, Grimm S, Ishida Y et al. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998;8:297–303.
Wong WW, Gentle IE, Nachbur U et al. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ 2010;17:482–487.
Zhao K, Song X, Huang Y et al. Wogonin inhibits LPS-induced tumor angiogenesis via suppressing PI3K/Akt/NF-kappaB signaling. Eur J Pharmacol 2014;737:57–69.
Yao J, Zhao L, Zhao Q et al. NF-kappaB and Nrf2 signaling pathways contribute to wogonin-mediated inhibition of inflammation-associated colorectal carcinogenesis. Cell Death Dis 2014;5:e1283.
He F, Wang Q, Zheng XL et al. Wogonin potentiates cisplatin-induced cancer cell apoptosis through accumulation of intracellular reactive oxygen species. Oncol Rep 2012;28:601–605.
Kim EH, Jang H, Shin D et al. Targeting Nrf2 with wogonin overcomes cisplatin resistance in head and neck cancer. Apoptosis 2016;21:1265–1278.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (no. 81300580, no. 81570623) and by Science and Technological Fund of Anhui Province for Outstanding Youth of China (grant number: 1608085J07).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
The authors determined the renoprotective effects of wogonin on cisplatin-induced acute kidney injury by targeting RIPK1-dependent necroptosis. Wogonin preserves, and even promotes, the tumor-killing effect of cisplatin in different types of tumor cells. Thus, wogonin should be further evaluated as a renoprotective adjuvant for kidney injury in cisplatin-treated cancer patients.
Rights and permissions
About this article

Cite this article
Meng, XM., Li, HD., Wu, WF. et al. Wogonin protects against cisplatin-induced acute kidney injury by targeting RIPK1-mediated necroptosis. Lab Invest 98, 79–94 (2018). https://doi.org/10.1038/labinvest.2017.115
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.115
This article is cited by
-
Cpd-A1 alleviates acute kidney injury by inhibiting ferroptosis
Acta Pharmacologica Sinica (2024)
-
The underlying mechanisms of cisplatin-induced nephrotoxicity and its therapeutic intervention using natural compounds
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
-
Generation of transgenic mice expressing a FRET biosensor, SMART, that responds to necroptosis
Communications Biology (2022)
-
Stratifin promotes renal dysfunction in ischemic and nephrotoxic AKI mouse models via enhancing RIPK3-mediated necroptosis
Acta Pharmacologica Sinica (2022)
-
Wogonin protects glomerular podocytes by targeting Bcl-2-mediated autophagy and apoptosis in diabetic kidney disease
Acta Pharmacologica Sinica (2022)

{kind=link}
{kind=link}