
Abstract
Notch signaling pathways have recently been implicated in the pathogenesis of metabolic diseases. However, the role of hepatic Notch signaling in glucose and lipid metabolism remains unclear and needs further investigation as it might be a candidate therapeutic target in metabolic diseases such as nonalcoholic steatohepatitis (NASH) and nonalcoholic fatty liver disease (NAFLD). We used hepatocyte-specific Notch1 knockout (KO) mice and liver biopsies from NASH and NAFLD patients to analyze the role of Notch1 in glucose and lipid metabolism. Hepatocyte-specific Notch1 KO mice were fed with a high fat diet (HFD) or a regular diet (RD). We assessed the metabolic phenotype, glucose and insulin tolerance tests, and liver histology. Hepatic mRNA expression was profiled by Affymetrix Mouse Gene arrays and validated by quantitative reverse transcription PCR (qPCR). Akt phosphorylation was visualized by immunoblotting. Gene expression was analyzed in liver biopsies from NASH, NAFLD, and control patients by qPCR. We found that Notch1 KO mice had elevated fasting glucose. Gene expression analysis showed an upregulation of glucose-6-phosphatase, involved in the final step of gluconeogenesis and glucose release from glycogenolysis, and perilipin-5, a regulator of hepatic lipid accumulation. When fed with an HFD KO mice developed overt diabetes and hepatic steatosis. Akt was highly phosphorylated in KO animals and the Foxo1 target gene expression was altered. Accordingly, a reduction in Notch1 and increase in glucose-6-phosphatase and perilipin-5 expression was observed in liver biopsies from NAFLD/NASH compared with controls. Notch1 is a regulator of hepatic glucose and lipid homeostasis. Hepatic impairment of Notch1 expression may be involved in the pathogenesis of human NAFLD/NASH.
Similar content being viewed by others


Main
The liver is an important organ for the regulation of energy homeostasis in mammals. Under fasting conditions, glycogenolysis and gluconeogenesis ensure energy abundance through the release of glucose into the circulation. Prolonged fasting initiates lipolysis including β-oxidation of fatty acids in hepatic mitochondria.1 In the fed state, glycogen synthesis and lipogenesis are consistently activated to store excess energy.2, 3 All these processes are controlled by hormonal, neuronal, and nutritional factors. The insulin pathway has a major role in the regulation of glucose absorption and uptake, glucose production, glycogen storage, and lipid/protein synthesis.4 Nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) are characterized by hyperinsulinemia caused by disturbance of regulatory mechanisms of energy homeostasis in the chronic state of overnutrition.5 The various cascades of deregulated signaling events leading to inappropriate energy storage in the liver remain incompletely understood. Recently, the canonical model assuming that phosphorylation of PKB/Akt is required for the regulation of hepatic glucose production has been questioned. Indeed, using an Akt1/Akt2/Foxo1 triple knockout (KO) mouse model, Lu et al6 provided evidence that hepatic glucose production is regulated independently of the Akt-dependent phosphorylation of Foxo1, whereas single KO of either Akt1 or Akt2 resulted in insulin resistance caused by a constitutive activation of Foxo1-dependent gene expression. In view of these findings, a re-evaluation of the current concept of hepatic glucose metabolism is needed and suggests that additional signaling pathways may be required.
The Notch pathway is an intercellular signaling cascade that has an important role in cell proliferation, differentiation, and fate determination. Thus, this pathway is essential for development and renewal of adult tissues.7 Notch1–4 are membrane-bound receptors that are activated by cell-to-cell interaction through their membrane-bound ligands (Jagged 1, -2, Delta-like ligand (Dll) -1, -3, -4). Their activation results in proteolytic cleavage and translocation of the Notch intracellular domain to the nucleus where it modulates transcription of target genes.8 Deletion of Notch1 during embryogenesis is lethal.9 Haploinsufficiency of Jagged 1 causes Alagille syndrome.10 Deregulation of Notch signaling has also been reported in hepatocarcinogenesis.11, 12 Recent works suggest a role for Notch signaling pathways in the regulation of metabolic processes. Changes in metabolism including body weight, energy metabolism, blood lipid composition, and expression of related genes have been observed in a model of mice haploinsufficient for the Notch ligand Dll-1.13 Elevated expression of Notch1 has been observed in small intestines of diabetic mice.14 Additionally, Notch activation and Notch inhibition have been reported to trigger hepatic steatosis and to reduce obesity, respectively, in mice fed with HFD.15, 16 Furthermore, regulation of lipid metabolism has been attributed to F-box and WD repeat domain-containing 7, a molecule that targets and degrades Notch.17 Finally, Notch1 has been reported to regulate Foxo1-mediated insulin sensitivity.18 In liver biopsies from NAFLD/NASH patients, the expression of Notch target genes HES1/HEY1 are elevated and correlated with insulin resistance and NAFLD activity score.19 These findings suggest a contribution of Notch signaling in the modulation of hepatic glucose and lipid metabolism and possibly NAFLD/NASH.
MATERIALS AND METHODS
Animals, Induction of Notch1 Deletion, Genotyping, and Southern Blot
Hepatocyte-specific Notch1 KO mice were generated as described previously.20 Briefly, Notch1lox/lox mice were crossed with AlbCre+/− mice on a C57Bl/6 background (Jackson Laboratory, Bar Harbor, ME, USA), expressing Cre recombinase under the hepatocyte-specific albumin promoter. Single transgenic AlbCre+/− mouse was used as control. Newborn mice were genotyped and the efficiency of Notch1 deletion was verified by Southern blot analysis using a Notch1-specific probe as described previously.21
Mice were maintained in the animal facility of the Department of Biomedicine at the University Hospital Basel, in a specific pathogen-free environment on a 12-h light and 12-h dark schedule. Food and drinking water were provided ad libitum. Experiments were approved by the veterinary office of Basel. Only adult (8-week-old) male mice were included in the study because of the higher prevalence of NALFD in males compared with that in females.
Patients
Liver biopsies from NAFLD, NASH, and (insulin-sensitive) control patients were obtained in the outpatient clinic of the Division of Gastroenterology and Hepatology, University Hospital Basel, Basel Switzerland. The use of biopsy material for this project was approved by the Ethics Committee of the Kantons Basel and Baselland, Switzerland. A part of the biopsy material that was not needed for routine histopathology was used for research purposes after obtaining written informed consent. Patients’ characteristics are summarized in Supplementary Table 1. NAFLD and NASH patients were classified based on the histology and the NAFLD activity score.
High Fat Diet Feeding Protocol
AlbCre Notch1 KO and AlbCre+/− (control) mice were fed with a high fat diet (HFD) containing 60% calories from fat (Laboratorio dottori piccioni srl, Gessate, Italy) or with a regular diet (RD) containing 10% calories from fat (Provimi Kliba Ag, Kaiseraugst, Switzerland). The composition of the diets is reported in Supplementary Table 3. Ten mice were used for each of the four feeding groups. Body weight was assessed weekly and fasting glucose level was measured every second week. Glucose and insulin tolerance tests were performed after 12 weeks of feeding. Mice recovered for 2 days and were fasted overnight before the liver was removed. Tissues were immediately snap frozen in liquid nitrogen and stored at −80 °C for further analysis.
Histologic Assessment of Liver and Pancreas Tissues
Liver and pancreatic tissues were fixed in 4% formaldehyde and processed for standard histologic assessment with H&E and Prussian blue staining. A blinded evaluation of liver histology was performed by an experienced hepatopathologist.
Fasting Glucose, Insulin, and HOMA-IR
Animals were fasted overnight and blood glucose level was measured using Ascensia Contour blood glucose monitoring system (Bayer AG Health Care, Zurich, Switzerland). Insulin was measured from 5 μl serum using Mercodia Ultrasensitive Mouse Insulin ELISA (Mercodia, Uppsala, Sweden). HOMA-IR was calculated based on the formula: fasting glucose (mmol/l)xfasting insulin (mIU/l)/22.5.22
Glucose and Insulin Tolerance Tests
For glucose tolerance test, mice were fasted overnight and a glucose solution was injected intraperitoneally at 2 g/kg of body weight. Glucose content was measured before and 15, 30, 45, 60, 90, and 180 min after glucose injection using Ascensia Contour blood glucose monitoring system. For insulin tolerance test, mice were fasted for 4 h and an insulin solution was injected intraperitoneally at 0.8 IU/kg of body weight. Glucose level was measured before and 15, 30, 45, 60, and 90 min after the insulin injection.
Microarray Analysis
Hepatic mRNA expression was profiled in three mice per group. Briefly, RNA from snap-frozen liver samples was extracted with Qiazol (Qiagen) and purified on RNeasy Mini Column Kit (Qiagen). Reverse transcription, second-strand synthesis, and in vitro transcription were performed according to the manufacturer’s instructions (Ambion). Samples were hybridized overnight to Affymetrix Mouse Gene ST 1.0 arrays (Affymetrix). Microarray analysis was carried out with Bioconductor packages of R statistical environment (http://www.R-project.org/). Normalization, background correction, and summarization on the level of transcript clusters were performed using RMA algorithm implementation of oligo package.23, 24 Genes were identified as differentially expressed if they showed a mean fold change above 2 with corresponding false discovery rate below 0.1.
Quantitative Reverse Transcription Polymerase Chain Reaction
One microgram of total RNA was reverse transcribed and quantitative PCR was performed using SYBR Green Polymerase Chain Reaction Master Mix (Applied Biosystems, Foster City, CA, USA). Intron-spanning primers were designed for each target gene and purchased at Microsynth (Balgach, Switzerland). All reactions were run in duplicate using a 7500 Fast Real-Time Polymerase Chain Reaction System (Applied Biosystems). Expression levels of the transcripts were normalized to Rpl19 or GAPDH using the ΔCt method. Primer sequences are reported in Supplementary Table 4.
Immunoblotting
Tissues were lysed in whole-cell lysis buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 10 mM PMSF, and 1 mM sodium orthovanadate. Lysates were cleared by centrifugation and protein concentrations were quantified by Bradford (Bio-Rad Protein Assay; Bio-Rad Laboratories AG, Reinach, Switzerland). Proteins were resolved by SDS-PAGE, transferred onto nitrocellulose membrane, and probed with specific antibodies. pS473-Akt (1:1000) and actin (1:1000) antibodies were from Cell Signaling (BioConcept, Allschwil, Switzerland) and Sigma-Aldrich Chemie GmbH (Buchs SG, Switzerland), respectively. Densitometry analysis of protein bands was performed with the ImageJ software (NIH Image).
Statistical Analysis
Mouse data are expressed as mean±s.e.m. and human data are shown as median. Comparisons were performed by the Student’s t-test or Mann–Whitney U-test using the GraphPad Prism 4.0 software. *P<0.05, **P<0.01, and ***P<0.001.
RESULTS
HFD Feeding Induces Overt Diabetes in AlbCre Notch1 KO Mice
We found that AlbCre Notch1 KO animals have a higher body weight than the control mice, suggesting that disruption of Notch1 signaling impairs body weight homeostasis (Figure 1a). The liver weight and the ratio liver/body weight remained unchanged in control and KO mice fed either with RD or HFD (Figures 1b and c). Interestingly, we did not observe any difference in the rate of body weight gain between the KO and the control animals fed with either RD or HFD (Supplementary Figure S1A). Furthermore, we did not measure an increased amount of food intake in the KO mice compared with control animals despite their initial higher body weight (Supplementary Figure S1B), suggesting that liver-specific Notch1 KO does not cause increased energy requirements.

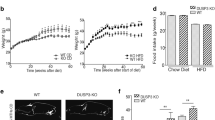
Hepatocyte-specific Notch1 knockout (KO) mice have elevated fasting glucose levels and are insulin resistant. Metabolic phenotype of 8-week-old male AlbCre Notch1 KO mice (KO) and AlbCre mice were used as controls. The animals (n=10 for each group) were fed with regular diet (RD) or high fat diet (HFD). (a) Body weight, (b) liver weight, (c) ratio of liver/body weight, (d) fasting glucose (mmol/l), (e) fasting insulin (mU/l), and (f) HOMA-IR (homeostatic model assessment Insulin resistance). Data are expressed as mean±s.e.m.
After feeding the animals with either RD or HFD for 12 weeks, we measured a significant weight gain in AlbCre Notch1 KO compared with control mice (Figure 1a). Interestingly, we noticed that KO mice fed with RD or HFD had significantly higher fasting glucose levels compared with control mice (Figure 1d).
Moreover, we show that the fasting glucose level measured after 12 weeks of feeding with HFD reached a pathological level (7.50±0.59 mmol/l) in the KO mice (Figure 1d). These data suggest an alteration in the hepatic glucose metabolism regulation in the KO animals. We then analyzed the fasting insulin levels and found that the level of insulin was increased in the groups fed with HFD. The mean insulin level was significantly higher in KO compared with control mice (Figure 1e). We did not observed any difference of the HOMA-IR between KO and control mice fed with RD. However, the HOMA-IR was significantly elevated in KO compared with control mice fed with HFD (Figure 1f), suggesting a key impact of the HFD. Taken together, these observations suggest a predisposition of the hepatocyte-specific KO modulating hepatic glucose metabolism towards the development of an insulin-resistant, diabetic phenotype in response to HFD.
Next, we analyzed the capacity of the mice to break down glucose by performing a glucose tolerance test on animals fed with RD or HFD for 12 weeks. As expected, we show significant higher blood plasma glucose content in Notch1 KO mice fed with HFD compared with the other groups, suggesting that these animals have impaired glucose tolerance (Figure 2a). We then tested the insulin sensitivity of Notch1 KO mice by performing an insulin tolerance test. We found that KO animals fed with HFD were insulin resistant compared with the mice from the other groups (Figure 2b). These data confirm deregulation of glucose homeostasis and suggest an alteration of the insulin action.

Hepatocyte-specific Notch1 knockout (KO) mice develop a diabetic phenotype in response to high fat diet (HFD) feeding over a 12-week period. (a) Glucose tolerance test was performed in 20-week-old hepatocyte-specific Notch1 KO and control mice after 12 weeks of feeding with HFD or regular diet (RD). Mice were starved for 14 h and 2 g/kg of glucose was injected intraperitoneally (n=10 for each group). Serum glucose levels were measured before, 15, 30, 45, 60, 90, 120, and 180 min after glucose administration. (b) Insulin tolerance test was performed within the same group of animals. Mice were starved for 4 h and 0.8 mU/kg of insulin was injected intraperitoneally. Serum glucose levels were measured before, 15, 30, 45, 60, 90, 120, and 180 min after insulin injection.
Glucose-6-Phosphatase and Perilipin-5 are Upregulated in Notch1 KO Mice
To study if the absence of Notch1 in the hepatocytes could alter the expression of genes that may favor gluconeogenesis, we compared hepatic gene expression between Notch1 KO and control mice fed with RD by performing transcriptome analysis. Interestingly, among the genes that are differentially changed, we measured an upregulation of hepatic glucose-6-phosphatase (G6PC), an important enzyme for the final steps of gluconeogenesis and glucose release from glycogenolysis, and perilipin-5 (PLIN5), a lipid droplet-associated protein known to regulate hepatic triglyceride accumulation, in Notch1 KO mice (Figure 3a). These results were then validated by quantitative reverse transcription PCR (RT-PCR) in a larger set of mice (Figure 3b).

Hepatocyte-specific Notch1 knockout (KO) mice have elevated expression of glucose-6-phosphatase and perilipin-5, and develop hepatic steatosis. (a) Microarray analysis was carried out in 3 out of 10 mice per regular diet (RD) feeding group. The heatmap shows the expression patterns of differentially expressed genes (fold change ≥2, false discovery rate ≤0.1) between Notch1 KO mice and control animals fed with a regular diet for 20 weeks. (b) Microarray results were validated by quantitative PCR (n=6 per group). Data are shown as mean±s.e.m. (c) Histology of the liver (hematoxylin and eosin (H&E) staining). Shown are representative images. (d) Microarray results showing the increased expression of three genes involved in lipid accumulation. Data are from three mice per group, fed with high fat diet (HFD). Results are shown as mean±s.e.m. (e) 3-Hydroxy-3-methylglutaryl coenzyme A reductase (HmgCoAR) expression level was measured by quantitative PCR in mice fed with RD (n=6 per group)
HFD Induces Increased Expression of Lipid Metabolism-Regulated Genes and Hepatic Steatosis in Notch1 KO Mice
Because PLIN5 is usually overexpressed in obese animals and is an important regulator of lipolysis, we performed histology analysis of liver sections from control and Notch1 KO mice fed with RD or HFD. We observed significant hepatic steatosis in 6 out of 10 Notch1 KO mice on HFD (Figure 3c and Supplementary Table S2). Only minor inflammation in 1 out of 10 Notch1 KO mice fed with HFD could be detected (Supplementary Table S2). Iron staining was negative in all animals. Pancreatic tissues appeared histologically normal without islet loss in randomly assigned mice of different feeding groups (data not shown). Our transcriptome analysis data revealed an elevated expression of Fas, 3-hydroxy-3-methylglutaryl coenzyme A reductase (HmgCoAR), and thyroid hormone responsive (Thrsp), three genes involved in the regulation of lipid metabolism, in Notch1 KO mice fed with HFD compared to control mice (Figure 3d). We then validated our results by quantitative PCR and showed that only HmgCoAR is significantly upregulated in Notch1 KO mice compared with control animals (Figure 3e).
Hepatic Akt is Phosphorylated in Notch1 KO Mice
It is well established that Akt negatively modulates the insulin-mediated transcriptional activity of Foxo1 leading to a reduction of gluconeogenic genes.6 As we have measured an increased expression of G6PC in Notch1 KO animals fed with RD, we therefore analyzed the phosphorylation status of hepatic Akt in control and Notch1 KO mice fed with RD or HFD. Surprisingly, we measured a higher magnitude of Akt phosphorylation in the liver of Notch1 KO mice fed with RD compared with control mice (Figures 4a and b). No difference was observed between the Notch1 KO and the control group when fed with HFD. We then analyzed the mRNA expression of two Akt-Foxo1 target genes, insig2 and VLDLR, in Notch1 KO and control mice. We expected to find a decrease of insig2 and an increase of VLDLR expression in NOTCH1 KO mice correlating with Akt phosphorylation. However, we found that insig2 and VLDLR expression levels increased and decreased, respectively (Figure 4c), suggesting that in our hepatocyte-specific Notch1 KO mouse, the defective modulation of hepatic glucose levels is independent of the classical Akt-Foxo1 signaling cascade.

Akt is phosphorylated in hepatocyte-specific Notch1 knockout (KO) mice and Foxo1-dependent target genes are differentially expressed. (a and b) Phosphorylated and actin were analyzed by western blotting and the signal intensity of the bands were quantified (n=10 per group). Results are expressed as mean±s.e.m. (c) Insig2 and Vldlr expression were assessed by microarray analysis in mice fed with regular diet (RD) (n=3 per group). Results are shown as mean±s.e.m.
Notch1 Expression is Reduced and G6PC/PLIN5 are Upregulated in Liver Biopsies From NAFLD/NASH Patients
Next, we quantified the mRNA expression of NOTCH1, HES1, HEY1, G6PC, and PLIN5 in liver biopsies from NAFLD/NASH patients and compared with normal liver samples. In line with our mouse data, we showed that Notch1 expression level is significantly reduced in NAFLD and NASH patients compared with control samples (Figure 5). We are aware that the reduction of NOTCH1 expression was only measured at the mRNA level and a quantification of NOTCH1 protein amounts would be more informative. However, no reliable antibodies against NOTCH1 are available presently. We also found that Notch1-target genes Hes1 and Hey1 levels are reduced in NAFLD and NASH patients compared with control samples. Finally, we reported that G6PC and PLIN5 transcripts expression were higher in NAFLD and NASH groups compared with the control group.

Hepatic NOTCH1 is reduced, and glucose-6-phosphatase (G6PC) and perilipin-5 (PLIN5) are upregulated in nonalcoholic fatty liver disease/nonalcoholic steatohepatitis (NAFLD/NASH) compared with control patients. The expression levels of NOTCH1, G6PC, PLIN5, HES1, and HEY1 were measured by quantitative PCR in NASH (n=17), NAFLD (n=13), and control (n=8) patients. Results are shown as boxplots.
DISCUSSION
In the present work, we identified hepatocyte-specific Notch1 KO mice as a murine model harboring an elevated expression level of G6PC and PLIN5 predisposes to the development of hepatic steatosis. Indeed, we show that Notch1 KO mice illustrate a phenotype susceptible to HFD-induced insulin resistance, diabetes and hepatic steatosis within 12 weeks. Although the mechanism associating Notch1 deletion in hepatocytes to transcriptional regulation of these genes remains unclear, our data suggest that it is independent of insulin-mediated Akt phosphorylation.
Presently, only few components of the Notch cascade have been studied in a different metabolic context. The complexity of phenotypic changes in Dll-1 haploinsufficient mice indicates an involvement of Notch signaling in many more biological processes than previously known. Indeed, Dll-1 haploinsufficiency in mice leads to decreased body weight, food consumption, and metabolized energy. A reduction of triglyceride and cholesterol levels in the liver as well as genes involved in cholesterol synthesis such as Idi1 and Hmgcs1 has also been observed.13 Taken together with our data, it suggests a regulatory role of Notch signaling in cholesterol and lipid biosynthesis, energy storage, and regulation of body weight. Combinatory possibilities of ligand-receptor binding and their various intra- and extracellular regulatory mechanisms are required to adjust context-specific distinct Notch-regulated gene expression patterns as during development. We hypothesize that similar specific ligand-receptor interactions and tissue-specific regulatory mechanisms might explain the observed differences on the phenotypes of Dll-1 haploinsufficient and Notch1 KO mice.
Pajvani et al18 have reported a direct binding of RBPJκ on the G6PC promoter modulating gluconeogenesis in a Foxo1-dependent manner. In their study, haploinsufficiency of Notch1 alone did not result in phenotypic changes. However, haploinsufficiency of both Notch1 and Foxo1 led to significantly higher insulin sensitivity compared with control mice. The hepatocyte-specific Notch1 KO model used in the present work shows clear phenotypic changes including hyperglycemia, hyperinsulinemia, body weight gain, and steatosis when fed with HFD. Thus, the cell-type-specific KO provides evidence for a primary hepatocyte-specific molecular change due to Notch1 deletion, which is most likely induces upregulation of hepatic glucose and lipid production as an origin for the observed systemic phenotypic changes. Although the data presented in this study seem controversial to the results reported by Pajvani et al,18 there are diverse possible mechanisms that could explain the discrepancy and that add new knowledge to the complexity of Notch signaling on hepatic glucose and lipid metabolism. First, the Notch1 haploinsufficiency model used by Pajvani has a reduction of Notch1 expression in all tissues, whereas Notch1 deletion is hepatocyte specific in our model. The concept that hepatic non-parenchymal cells could be effectors of metabolic syndrome and insulin resistance has been discussed previously.25 Indeed, these non-parenchymal cells seem to be not only bystanders but can react actively in response to changes of their environment. Thus an alteration of Notch signaling in hepatocytes might consequently trigger a compensatory mechanism in non-parenchymal cells that leads to an elevated glucose and lipid production. Indeed, α-smooth muscle actin staining of liver sections from control and KO mice showed a reduction of activated hepatic stellated cell (HSC) phenotype in hepatocyte-specific Notch1 KO animals, suggesting a switch towards fat-storing phenotype of HSCs (Supplementary Figure S2). A more in-depth analysis of Notch signaling in parenchymal vs non-parenchymal cells in the context of insulin resistance and steatosis is needed to clarify this issue. Second, the model used by Pajvani is a combined haploinsufficiency of Foxo1 and Notch1. It is well known that Foxo family members are important effectors of insulin-mediated hepatic glucose production.26 Therefore, the absence of Foxo1 in their model might lead to a reduction of gluconeogenesis. Third, Valenti et al19 have reported that the canonical Notch targets, HEY1 and HES1, are upregulated in NAFLD patients, suggesting a role for Notch signaling in insulin resistance and steatosis. In our NASH and NAFLD cohorts, HES1 and HEY1 expression levels are reduced compared with the control group. Furthermore, in our hepatocyte-specific KO mice, Hey1/Hes1 are not differentially expressed (data not shown). Our data suggest a noncanonical Notch signaling-mediated G6PC and PLIN5 upregulation. Further analysis of cell-type-specific expression of Notch receptors and ligands is needed to clarify the underlying mechanism.
The canonical model of hepatic glucose regulation in which Akt is required to modulate Foxo1 transcriptional activity has been discussed.6 Indeed, it has been reported that the regulation of hepatic glucose production in Akt1/Akt2/Foxo1 triple KO mice was comparable to control animals, suggesting that this process is independent of Akt-Foxo1 signaling. In our model of hepatocyte-specific Notch1 KO, we measured a higher magnitude of Akt activation in the KO mice compared with the control animals (Figures 4a and b), resulting in an alteration of Foxo1 target genes expression (Figure 4c), probably due to Foxo1 nuclear exclusion and degradation. Therefore, a reduction of G6PC expression was expected in Notch1 KO animals. Surprisingly, we measured an increased expression of G6PC in KO mice (Figure 3b). These data suggest that G6PC expression in hepatocyte-specific Notch1 KO mice is independent of Akt-Foxo1 signaling.
Insulin resistance predisposes to hepatic steatosis caused by an increase in lipolysis in adipose tissues and enhanced flux of free fatty acids to the liver.27 Insulin resistance is generally associated with the expression of lipid-activated transcription factor PPARα, which transcriptionally increases PLIN5 expression. It is well documented that PLIN5 contributes to lipid accumulation by altering lipid droplet metabolism through inhibition of triglyceride degradation and/or reducing fatty acid oxidation.28 In our murine model of hepatocyte-specific Notch1 deficiency, insulin resistance and hepatic PLIN5 upregulation were measured and might contribute to the lipid droplet accumulation observed.
In summary, our data confirm the proposed role of Notch signaling in metabolism. We provide new evidence for the Notch1-dependent regulation of hepatic glucose and lipid production and thus propose Notch1 as a new important player in the pathophysiology of NAFLD/NASH. Although the full molecular mechanism remains unclear, the data suggest a more complex regulatory mechanism involving distinct Notch ligands and receptors on parenchymal and non-parenchymal cells in the liver. A detailed analysis of cell-type-specific ligand binding of Notch1 as well as Notch1/RBPJκ transcriptional regulation of genes involved in hepatic metabolism are needed to evaluate the potential future drug targets for the treatment of insulin resistance and hepatic steatosis.
References
Rui L . Energy metabolism in the liver. Compr Physiol 2014;4:177–197.
Markan KR, Jurczak MJ, Brady MJ . Stranger in a strange land: roles of glycogen turnover in adipose tissue metabolism. Mol Cell Endocrinol 2010;318:54–60.
Wong RH, Sul HS . Insulin signaling in fatty acid and fat synthesis: a transcriptional perspective. Curr Opin Pharmacol 2010;10:684–691.
Newsholme P, Cruzat V, Arfuso F et al. Nutrient regulation of insulin secretion and action. J Endocrinol 2014;221:R105–R120.
Sookoian S, Pirola CJ . NAFLD. Metabolic make-up of NASH: from fat and sugar to amino acids. Nat Rev Gastroenterol Hepatol 2014;11:205–207.
Lu M, Wan M, Leavens KF et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med 2012;18:388–395.
Aster JC . In brief: Notch signalling in health and disease. J Pathol 2014;232:1–3.
Hori K, Sen A, Artavanis-Tsakonas S . Notch signaling at a glance. J Cell Sci 2013;126 (Part 10):2135–2140.
Jandke A, Da Costa C, Sancho R et al. The F-box protein Fbw7 is required for cerebellar development. Dev Biol 2011;358:201–212.
Boyer-Di Ponio J, Wright-Crosnier C, Groyer-Picard MT et al. Biological function of mutant forms of JAGGED1 proteins in Alagille syndrome: inhibitory effect on Notch signaling. Hum Mol Genet 2007;16:2683–2692.
Dill MT, Tornillo L, Fritzius T et al. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology 2013;57:1607–1619.
Villanueva A, Alsinet C, Yanger K et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 2012;143:1660–1669, e1667.
Rubio-Aliaga I, Przemeck GK, Fuchs H et al. Dll1 haploinsufficiency in adult mice leads to a complex phenotype affecting metabolic and immunological processes. PLoS One 2009;4:e6054.
Min XH, Yu T, Qing Q et al. Abnormal differentiation of intestinal epithelium and intestinal barrier dysfunction in diabetic mice associated with depressed Notch/NICD transduction in Notch/Hes1 signal pathway. Cell Biol Int 2014;38:1194–1204.
Bi P, Shan T, Liu W et al. Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat Med 2014;20:911–918.
Fowler JC, Zecchini VR, Jones PH . Intestinal activation of Notch signaling induces rapid onset hepatic steatosis and insulin resistance. PLoS one 2011;6:e20767.
Onoyama I, Suzuki A, Matsumoto A et al. Fbxw7 regulates lipid metabolism and cell fate decisions in the mouse liver. J Clin Invest 2011;121:342–354.
Pajvani UB, Shawber CJ, Samuel VT et al. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat Med 2011;17:961–967.
Valenti L, Mendoza RM, Rametta R et al. Hepatic notch signaling correlates with insulin resistance and nonalcoholic fatty liver disease. Diabetes 2013;62:4052–4062.
Dill MT, Rothweiler S, Djonov V et al. Disruption of Notch1 induces vascular remodeling, intussusceptive angiogenesis, and angiosarcomas in livers of mice. Gastroenterology 2012;142:967–977, e962.
Radtke F, Wilson A, Stark G et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 1999;10:547–558.
Matthews DR, Hosker JP, Rudenski AS et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412–419.
Carvalho BS, Irizarry RA . A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010;26:2363–2367.
Irizarry RA, Bolstad BM, Collin F et al. Summaries of Affymetrix GeneChip probe level data. Nucleic acids Res 2003;31:e15.
Leclercq IA, Da Silva Morais A, Schroyen B et al. Insulin resistance in hepatocytes and sinusoidal liver cells: mechanisms and consequences. J Hepatol 2007;47:142–156.
Gross DN, van den Heuvel AP, Birnbaum MJ . The role of FoxO in the regulation of metabolism. Oncogene 2008;27:2320–2336.
Schenk S, Saberi M, Olefsky JM . Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 2008;118:2992–3002.
Greenberg AS, Coleman RA, Kraemer FB et al. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest 2011;121:2102–2110.
Acknowledgements
This work was supported by SNF Score Grant 32323B_123815 and Stiftung Propter Homines grant to DS.
Author contributions
CB, MTD, AP, ZM, IK, GM, LT, and FHTD performed the experiments and analyzed the data. CB, MTD, DS, MHH, and FHTD designed the experiments. CB, MTD, ZM, GM, FM, and FHTD wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
The implication of Notch signaling in lipid and glucose metabolism is unclear. The authors show that Notch1 depletion impairs glucose-6-phosphatase and perilipin-5 expression and is associated with diabetic phenotype and hepatic steatosis. These results confirm the metabolic role of Notch1 and suggest Notch1 as a new player in the physiopathology of non-alcoholic steatohepatitis and non-alcoholic fatty liver disease.
Rights and permissions
About this article

Cite this article
Bernsmeier, C., Dill, M., Provenzano, A. et al. Hepatic Notch1 deletion predisposes to diabetes and steatosis via glucose-6-phosphatase and perilipin-5 upregulation. Lab Invest 96, 972–980 (2016). https://doi.org/10.1038/labinvest.2016.75
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2016.75
This article is cited by
-
Exposure to PM2.5 affects blood lipid levels in asthmatic rats through notch signaling pathway
Lipids in Health and Disease (2019)

{kind=link}
{kind=link}