
Abstract
Chronic traumatic encephalopathy (CTE) has been in the medical literature since the 1920s. It is characterized clinically by diverse neuropsychiatric symptoms, and pathologically by variable degrees of phosphorylated tau accumulation in the brain. The evolving paradigm for the pathogenesis of CTE suggests that concussion or subconcussion from athletic participation initiates a cascade of pathologic events, encompassing neuroinflammation and protein templating with trans-synaptic neurotoxicity. The end result is neurologic and neurobehavioral deterioration, often with self-harm. Although these concepts warrant further investigation, the available evidence permits no conclusions as regards the pathogenesis of the reported findings. Investigations into the role of premorbid or co-morbid neurodegenerative diseases has been limited to date, and in-depth genetic analyses have not been performed. The role of concussion or subconcussion if any, whether and how the condition progresses over time, the extent of phosphorylated tau in clinically normal athletes, the role of phosphorylated tau as a toxic species versus an inert disease response, and whether protein templating has any in vivo relevance remain to be elucidated.
Similar content being viewed by others

Main
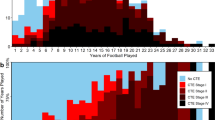
Chronic traumatic encephalopathy (CTE) is the accepted term for a pattern of phosphorylated tau (ptau) deposition in the brain that appears to differ from age-related accumulations and neurodegeneration.1 According to data from the largest CTE series to date, ptau tends to occur as localized accumulations in depths of sulci and perivascular areas of the cerebral cortex, particularly frontal, temporal, and insular cortices. CTE further tends to involve cortical laminae 2 and 3, relative to AD and aging where ptau predominates in laminae 3 and 5. Extensive medial temporal lobe involvement, and involvement of the brainstem tegmentum, may also be present. Axonal varicosities in the deep cortex and subcortical white matter are variously described. The gross brain varies from markedly atrophic to normal, while cavum septum pellucidum and septal fenestrations are common findings. Staging schemes have been proposed in recent years, in an attempt to understand the kinetics of the process as a function of time and clinical manifestations.1, 2
CTE is associated with contact sports and is often considered a variation of so-called dementia pugilistica (DP),3, 4, 5 a long known condition in boxers associated with neurologic decline and neurofibrillary degeneration at autopsy.6 Although generally accepted as a distinct entity, DP has been controversial since its original description, with absence of prospective data,7 surprisingly few studies with autopsy correlation5, 8, 9, 10, 11, 12, 13 and lack of accounting for co-morbidities such as substance abuse, infection, and vascular or neurodegenerative disease.13 In the only large-scale study of boxers to date, Roberts14 investigated 250 boxers from a cohort of 16 781 boxers in the UK and found 37 with neurological lesions, suggesting an overall prevalence of 17%. Some differences between CTE and DP have been suggested in a recent review, including clinical presentation, age at onset, association with APOE genotype,8 and tendencies for neurological versus psychiatric signs, although none of these features provide a clear separation. Both CTE and DP tend to be viewed as variants of the same condition—progressive tauopathy caused by brain trauma.1
The exposure to sport along with the increase in ptau in parenchymal brain tissue has suggested head trauma as the underlying biomechanical etiology of CTE. Indeed, the recent, heightened awareness of concussion and subconcussion as potentially important15 comes from studies in National Football League (NFL) players. Rare cases of CTE have been suggested in hockey player,1 professional wrestlers, rugby players,16 soccer players,12 a professional baseball player, and a circus performer.17 The issue of CTE possibly resulting from combat-related traumatic brain injury (TBI) has also been raised; however, its existence as an entity in combat veterans and potential mechanisms of injury, have yet to be confirmed.18
The clinical manifestations associated with CTE in recently characterized cases are largely psychiatric, and include aggression, explosive anger, impaired impulse control, domestic disarray, depression, and heightened suicidality,1 although cognitive impairment, short-term memory loss, and headache are also reported. The nature and rate of progression, however, remains an open question.8
ACCEPTED CTE PARADIGM
The gaps in the knowledge of CTE are substantial, and the collective human data, which are retrospective, and largely based on self-selected cases, permit no conclusions as yet, regarding etiology or its existence as a distinct clinicopathologic entity. Nevertheless, the CTE paradigm from TBI to neurodegeneration is generally accepted, with efforts directed more at identifying the molecular events responsible for the neurodegeneration, than confirming its existence.19
TBI and Concussion
The paradigm begins with the heterogeneous, imperfectly modeled, and complex condition termed TBI. In-depth reviews of TBI are available.19 Briefly, TBI most often signifies loss of consciousness and is arbitrarily termed ‘mild’ if the loss of consciousness is up to 30 min. Alteration in consciousness for up to 24 h, or posttraumatic amnesia for up to 24 h, is also accepted under the mild TBI umbrella. The diagnosis of mild TBI may be entirely subjective, as it is often based on self-reported neurological symptoms.20 Thus, although mild TBI is portrayed in the literature as a definable condition, it encompasses a wide spectrum of potential biomechanical precursors, including nature and type of impact, directionality of acceleration-deceleration phenomena, and individual susceptibilities, as well as the interpretation itself which is often subjective, and often provided by physicians and other personnel with wide variability in experience in the diagnosis of TBI. There is a tendency for ‘TBI’ to be used for military-related TBI, given the spectrum from mild to severe, and for ‘concussion’ to be used in sport, in place of mild TBI.
Concussion in contact sports, either objective or subjective, is a common and inevitable accompaniment of a range of sports.21 Indeed, in high school sports in the US, the concussion rate in girls’ soccer is comparable with that of boys’ football.22 The diagnosis of concussion often presents a challenge among sports medicine physicians and athletic trainers, just as mild TBI may be a challenge to the medical personnel in armed conflicts. Assessment by physicians with specific expertise in concussion is ideal, although this is often not available. Codified evaluation and management strategies are in progress.21
Risk factors for concussion have been only obliquely addressed in the context of CTE. Among these are history of previous concussion, number and severity of concussion, age, gender, pre-existing mood disorders, pre-injury learning disabilities such as attention deficit hyperactivity disorder, and history of migraines.23 Prolonged concussive symptoms or post-concussive syndrome that may persist in a minority of patients for weeks or even years, adds an additional level of complexity and pathophysiological uncertainty to the concept of TBI.
It is of some passing interest that one high-profile NFL football player who was determined to have some degree of ptau deposition post-mortem had no documented concussions during his football career at any level prior to committing suicide at the age of 43. This case tends to de-emphasize the importance of concussion per se, and elevates subconcussive impact as a potential etiological factor. Regardless of the specific concussion history in athletes, however, uncertainties regarding concussion and potential biomechanical antecedents to CTE are evident, notwithstanding the certainty with which concussions are viewed as etiological in the media as well as the medical literature.24
Diffuse Traumatic Axonal Injury (DAI) and Biomechanics of Concussion
Understanding concussion in the acute state from the standpoint of neuropathology is problematic in that the neurological deficit is transient and without mass effect, ie, the patients survive and do not require neurosurgery, precluding pathological evaluation. The pathology and mechanics of concussion are therefore difficult to study in vivo. One study that looked at brain changes in individuals who expired from other causes shortly after concussion found evidence of axonal injury, including involvement of the fornix, suggesting similarities with DAI and involvement of memory circuitry.25 Such data, however, are sparse. On the other hand, studies on brain contusion, a commonly observed lesion and one that is definable based on anatomic pathology have led to the basic concept that sheer stresses, or the movement of one tissue plane over another, are necessary for parenchymal brain injury.26 As emphasized by Holbourne27 more than 70 years ago, the brain’s relative incompressibility and lack of rigidity necessitate shear stresses over compressive stresses and rotational acceleration over linear acceleration. One could reasonably speculate, therefore, that shear stresses, rotational acceleration, and axonal disruption or injury are the basic physical precursors to concussion.
Purely biomechanical models in primates from the 1970s and 1980s may have also shed some initial light on concussion indirectly through the characterization of DAI. In these early primate models, it was determined among other factors that acceleration in the coronal plane28 and low strain rate (prolonged interval over which acceleration occurs) favored prolonged traumatic unconsciousness, poor outcome, and DAI at necropsy.29, 30 The biomechanics of DAI may therefore follow that of concussion, albeit with more severe clinical and pathological outcome. This is also consistent with the clinical definition of DAI requiring 6 h of traumatic unconsciousness,31 which differs from concussion largely on the basis of duration of the unconsciousness.
At the experimental level, the last 30 years has seen a proliferation of in vitro32 and mammalian33 trauma models, which have led to an exponential expansion of data implicating essentially all major molecular disease mechanisms. The collective data indicate that TBI and otherwise biomechanical forces acting on parenchymal brain tissue result in pleiotropic deleterious, biochemical sequelae, encompassing signal transduction, elaboration of toxic proteins, unfolded protein responses and ER stress, oxidative stress, dysfunction in mitochondria and energy metabolism, channelopathy effects with elaboration of pore forming molecular complexes, inflammatory cytokine production, perturbations in calcium and electrolyte metabolism, and induction of apoptosis, among other mechanisms.34, 35, 36, 37, 38, 39, 40, 41 With respect to CTE, however, these processes lead to, or otherwise facilitate, tau phosphorylation via altered kinase-phosphatase metabolism,42 resulting in microtubule instability and precipitation of ptau as toxic, insoluble intraneuronal and intra-astrocytic inclusions.43
CTE as a Prion Disease
Somewhat concerning are the studies suggesting protein templating with trans-synaptic transmission44 and the incorporation of the tauopathies in the lexicon of prion (or prion-like) diseases. Transgenic mice overexpressing P301L tau in the entorhinal cortex, for example, demonstrate de novo wild-type ptau in brain regions synaptically connected with the performant pathway.45 Further, prion-like self-propagating ‘strains’ of tau appear along neuroanatomical pathways following intracerebral inoculation of experimental mice transgenic for human tau.46 Similar strain propagation of amyloid-β has been demonstrated with synthetic peptides, and suggested as basis for phenotypic variability to human AD.47, 48 These data taken together make a sophisticated case for spreading toxicity in a prion-like manner.
Phosphorylated tau species according to this hypothetical paradigm thus ‘spreads’ along neuroanatomical pathways, leading to disease progression and neurodegenerative disease. The frequent involvement of frontal and temporal lobes by the neurotoxic process is said to cause neurobehavioral symptoms such as disordered impulse control, explosive aggressiveness, extreme impulsivity, impaired judgment and social function, and heightened suicidality.1 Medial temporal lobe involvement may disrupt episodic memory, whereas the involvement of the brainstem implicates a number of functions.49
In short, TBI induces neuroinflammation, leading to tau phosphorylation, leading in turn to disease progression, possibly encompassing unfolded protein responses, ptau templating with trans-synaptic spread of toxic, self-propagating strains, and neurodegeneration in a distribution that favors neuropsychiatric disturbances, impaired cognition, and motor signs (Figure 1). Moreover, head trauma, even singular head trauma, may initiate this process according to reports,2 and may set up a cellular milieu favoring neurodegeneration, including AD, frontotemporal lobar degeneration, and amyotrophic lateral sclerosis.

The empirical data regarding tauopathy and contact sports appear to indicate that certain contact sports may be associated with an altered distribution of phosphorylated tau when examined at autopsy years to decades following sports participation. The significance of this change and the correlation between such changes and clinical signs are matters of considerable uncertainty, as more research with more rigorous prevalence data is necessary even for this preliminary assertion. This is in contrast, however, to the accepted paradigm, none of which has been objectively demonstrated in humans. The accepted paradigm indicates that head trauma per se and in particular concussion, initiates the overall process, which in turn sets in motion neuroinflammatory processes that span a large spectrum of biology. Phosphorylation of tau via pathological alteration of kinase-phosphatase equilibrium subsequently occurs in brain regions vulnerable to mechanical stress. Soluble, low-n phospho-tau oligomers of altered conformation or ‘strain’ then spread along neuroanatomical pathways, effect conformational changes in other phospho-tau species via protein templating, and cause trans-synaptic neurotoxicity, which fosters disease progression. The tendency for frontotemporal involvement by phosphorylated tau further is suggested to be the basis for complex behaviors, such as impulse control, mood, and attention, as well as the reported psychiatric manifestations of CTE. Involvement of memory pathways is suggested to disrupt episodic memory. The overall neurotoxic cascade is said to overlap with, include, or even cause, pathology of Alzheimer’s disease, frontotemporal lobar degeneration, and/or amyotrophic lateral sclerosis.
PROBLEMS WITH THE CURRENT PARADIGM
The DP Literature
The concept for present day CTE, including the naming of the condition, is based on DP, which, while broadly accepted as a clinicopathological entity, is comprised of a relatively small number of cases with remarkably heterogeneous pathology. Moreover, the few DP cases described in the literature have been examined over many decades using differing techniques, including dyes and silver impregnation, with a minority of cases assessed via immunohistochemistry for ptau and amyloid-beta.
The term ‘punch drunk’ appeared in the medical literature in a 1928 JAMA article by Martland, in which small hemorrhages were emphasized pathologically. The first case of DP with neurofibrillary degeneration was described by Brandenberg and Hallervorden50, in a 51-year-old retired boxer (retired at age 28 after 11 years as an amateur boxer), although plaque pathology and cerebral amyloid angiopathy were also noted, as was death from intracerebral hemorrhage, raising the possibility of early onset AD. Courville51 in 1962 reported an autopsy case of punch drunk syndrome, although no neurofibrillary degeneration or features presently ascribed to CTE were mentioned. Constantinides and Tissot in 196752 described severe degeneration of the substantia nigra with numerous neurofibrillary tangles in a 58-year-old man who had been retired from boxing for 34 years,5 raising the possibility of co-morbid tauopathy. Payne11 in 1968 described autopsy findings in six boxers in their forties, calling attention to septal abnormalities. CTE changes according to modern concepts (perivascular and superficial neurofibrillary change, neurofibrillary change in the depth of sulci) were not apparent, although ‘early’ neurofibrillary changes were observed in two brains, and were considered non-specific.
The largest series to date, however, was that of Corsellis et al,5 who described findings in 15 boxers. In this seminal article, neurofibrillary degeneration out of proportion to plaque pathology was established as an integral pathological change, while also emphasizing septal changes, substantia nigra degeneration, and cerebellar scarring. Clinical findings often included speech abnormalities, ataxia, and movement disorders. A closer look at the cases, however, indicates a level of complexity and variability. At least 6 of the 15 cases were accompanied by heavy alcohol use. Co-morbidities such as hypertensive vascular disease with lacunar infarcts were evident in some cases. One patient had tabes dorsalis, another had a cavernous malformation of the globus pallidus, while others had little pathology and were neurologically normal during life. A number of the subjects boxed in the early part of the twentieth century, when several hundred fights were not uncommon. The reduced exposure in boxers presently may significantly reduce the risk of chronic neurologic sequelae.53 Also noteworthy is the subsequent examination of many of the original cases using immunohistochemistry to amyloid beta, which showed not only diffuse plaques, but also neuritic plaques which in some cases were sufficient in quantity to warrant the diagnosis of AD.9, 54 Extreme substantia nigra degeneration with neurofibrillary change in residual neurons also raises the possibility of sporadic tauopathy (e.g., corticobasal syndrome, progressive supranuclear palsy). Atrophy involving hypothalamus and mammillary bodies in some of these cases warrants discussion of thiamine deficiency and Wernicke–Korsakoff syndrome. It should also be noted, in light of the recently identified genetic lesions that cause FTLD/ALS, family history was not commented upon in any of the cases in this series. Thus, although the description of neurofibrillary degeneration was remarkable, the limited numbers, extensive head trauma exposure, and co-morbidities indicate a level of uncertainty even in the relatively well-accepted entity of DP.
More recent cases are fewer but perhaps more compelling. For example, Hof et al55 described neurofibrillary tangle clusters in frontal and temporal cortices in a 24-year-old autistic patient who was prone to frequent and protracted self-injurious head-banging. The findings included a tendency for focal involvement of cortical laminae 2 and 3, now considered a feature of CTE. Geddes et al12 noted neurofibrillary change with a tendency for basal cortical involvement by tau immunohistochemistry in two boxers in their twenties, and further called attention to perivascular tau, also considered a hallmark of CTE. Neither subject had clinical signs suggesting DP, however. About the same time, the New England Journal of Medicine presented a case of multisystem neurodegenerative disease, including involvement of the spinal cord and substantia nigra, in a 67-year-old retired boxer (10 year career with over 100 bouts).56 Interestingly, probable amyotrophic lateral sclerosis was diagnosed in the patient’s brother, raising the possibility of pathogenic mutation. Schmidt et al57 examined brain tissue from this same patient and one additional retired boxer, aged 78, and noted that the tau isoform profile in soluble fractions resembled that of AD. The second patient, however, had Lewy bodies in the substantia nigra, again raising the issue of co-morbid neurodegenerative disease. Areza-Fegyveres et al58 reported a case of DP with a clinical progression that was indistinguishable from AD. He had fought more than 60 bouts but had never been knocked out.
McKee and colleagues on the other hand more than doubled the cases of sport-associated tauopathy reported in the literature with a remarkable brain procurement effort, particularly targeting NFL athletes.1 Emphasis was placed on the pattern of tauopathy, best illustrated by thick, sledge microtome whole-mount immunostains. Thus, the updated pathology of CTE includes: (i) localized neuronal and glial accumulations of phosphorylated tau involving perivascular areas of the cerebral cortex, sulcal depths, and with a preference for neurons within superficial cortical laminae; (ii) multifocal axonal varicosities involving deep cortex and subcortical white matter; (iii) relative lack of Aβ deposits; and (iv) TDP-43-positive inclusions and neurites. The first of the above findings is arguably the most robust, as these distributions are generally not described as incidental accumulations with age or as a prominent feature in AD. Axonal varicosities, relative lack of Aβ, and TDP-43 pathology may be difficult to distinguish from other diseases and controls in blinded analyses.
In a review of the CTE (including DP) literature to date, however, Gardner et al8 noted that of the 61 pure athlete cases reported by McKee et al, 25% demonstrated no pathological features of CTE, and 25% had ‘pure’ CTE pathology. Of those with pure CTE pathology, clinical findings were sometimes absent or nonspecific. Noteworthy also was that only a minority of cases showed disease progression, contrary to the commonly held view that CTE is a neurodegenerative disease.
Both DP as classically defined, and modern day CTE, therefore span a wide spectrum from normal to advanced disease, from the standpoint of both clinical presentation and neuropathological findings, with disease progression being doubtful in many cases according to one review. The broad question of whether DP or CTE merits inclusion in the broad category of neurodegenerative disease may be debated in light of these data, although the low thresholds for exposure and diagnosis are understandable as information is still accumulating.
Ptau as a Mediator of Disease
Ptau accumulation occurs in a long list of conditions and is manifestly downstream in Alzheimer’s disease according to the amyloid cascade hypothesis.59 Some have suggested that ptau is not only a reactive phenomenon but possibly even a beneficial or adaptive disease response.60 Neurofibrillary degeneration, for example, is associated with adducts of advanced glycation61 and lipid peroxidation;62 these chemical modifications contribute to protein insolubility, and target inert accumulations for degradation. Ptau has been shown to sequester toxic-free radicals and heavy metals,63 and is known to accumulate in viable cells for decades.64 Transgenic tauopathy models have further demonstrated lack of neurotoxicity associated with ptau.65 The appearance of phosphorylated tau in areas of biomechanical stress or otherwise neuroinflammation (e.g., depths of sulci, perivascular areas) may therefore be less indicative of toxicity than of reactivity.
The kinetics of ptau accumulation during life is difficult to monitor, although research into tau-targeted positron-emission tomography is progressing,66 and may provide insight into ptau progression over time, i.e., whether it progresses, stabilizes, or even regresses. The known age-related ptau accumulations will require rigorous attention to variations of normal, particularly in light of the recently described, but reportedly very common, primary age-related tauopathy.67 In this aging pattern, ptau involvement of anterior-medial temporal lobe and brainstem tegmentum, often emphasized in CTE, is an expected finding.68, 69
It should also be noted that the prion-like, tau templating concept, while raising novel issues about basic protein chemistry, confronts the shortcomings of the in vitro experimentation when applied to human disease, such as (i) requirement of one to several, pathogenic mutations; (ii) utilization of supraphysiologic concentrations of mutated or insoluble, phosphorylated protein; (iii) variable pathology; and (iv) variable behavioral correlates.45, 70, 71 The juxtaposition of two pieces of human data may also cast doubt on ptau templating as an in vivo pathophysiologic event. Braak et al69 note that ptau, as detected by AT8 immunohistochemistry, is found earliest in the locus ceruleus, and as early as the first decade of life. In turn, the locus ceruleus is said to be ‘unsurpassed’ in the diffuseness and ubiquity of its connections throughout the nervous system.72 If ptau templating, cell-to-cell transmission, and spreading toxicity in a prion-like manner were in vivo phenomena, it is reasonable to suggest that clinically significant neurodegeneration would occur earlier in life and with greater regularity. Nevertheless, protein templating is increasingly accepted as an in vivo occurrence and has led to the palpable fear that a neurodegenerative process could result from a single blow to the head (http://www.psychologytoday.com/blog/invisible-wounds/201208/disturbing-new-study). This appears unwarranted from the standpoint of human data.
Genetic Predisposition to Chronic Neurodegeneration in Professional Athletes
It is commonly stated that genetic susceptibility (yet to be clarified) may underlie the sporadic appearance of CTE in athletes.16 In the case of American football, however, CTE is reported in essentially all professional athletes examined to date. In the absence of subclassification, such a high percentage precludes genetic susceptibility studies, as no one is genetically resistant. That said, apolipoprotein E genotype (APOE) is often discussed, given its role as the major genetic risk for sporadic AD. In the largest series to date, APOE allelic frequencies were comparable with the general population.1 An overrepresentation of the ɛ4 allele in boxers with neurological impairment was noted in one study, although autopsy findings were not available for correlation.73
Relevant to the discussion going forward may be the expanding list of pathogenic mutations and polymorphisms in frontotemporal dementia (FTD) and ALS,74, 75 particularly because (i) signs of FTD overlap with those reported in CTE; (ii) FTD typically presents in middle age; and (iii) co-occurrence of ALS and CTE is reported in some cases.1 FTD is also the second most common type of presenile dementia after AD and, importantly, the percentages of FTD and ALS patients with known pathogenic mutations have increased substantially in recent years.74 Among genetic variants, microtubule-associated protein tau (MAPT) and progranulin (GRN) mutation tend to favor an FTD presentation, while superoxide dismutase-1 (SOD1) mutations present as ALS.76 Mutations involving C9ORF72, TAR DNA-binding protein-43 (TARDP), Valosin-containing protein (VCP), p62/sequestosome-1 (SQSTM1), and ubiquilin 2 (UBQLN2), may have mixed FTD-ALS phenotypes.
C9ORF72 mutation involves expanded hexanucleotide repeats on the non-coding portion of chromosome 9, and is now the most common mutation in both ALS and FTD, accounting for up to 46% and 29% of familial ALS and FTD cases, respectively, in European populations.77 This recent finding may add to the understanding of CTE in that a subset of athletes presenting with signs of FTD and/or ALS may be excluded from the CTE category by genetic analysis. It also raises the issue of potential outcome in athletes with unrecognized genetic susceptibility to known diseases, ie, whether athletic participation influences neurodegeneration or is otherwise incidental in individuals with expanded C9ORF72 repeats.
The assignment of TDP-43 immunohistochemistry to the cardinal pathological features of CTE may be problematic owing to morphologic heterogeneity of TDP-43 immunoreactivity apparent in a range of conditions. TDP-43 positivity is found to varying extents with age and AD.78 In the FTD/ALS spectrum, TDP-43 positivity may be present in variable cell types (neurons versus glia), subcellular compartments (nucleus versus cytoplasm), distributions (superficial cortical versus diffuse versus subcortical), and morphologic expressions (perikaryal and nuclear inclusions of various morphologies versus neurites).79 TDP-43 positivity is further observed in a subset of familial and sporadic FTD, with and without TARDP, GRN, and C9ORF72 mutation.74 The finding in CTE cases is therefore not surprising and appears to be an empirical phenomenon, rather than a pathogenic signature. Rigorous controls for age and other competing diagnoses are probably necessary before accepting TDP-43 reactivity as hallmark pathology.
MAPT mutation also may be relevant to CTE given that both FTD with MAPT mutation and CTE show increased ptau at autopsy, both are reported to show behavioral disturbances, and both are reported to show cognitive deterioration and parkinsonism, in at least some cases.74 MAPT mutations account for 5–20% of familial FTD cases, which is significant given the tendency for pathogenic mutation in FTD overall. MAPT mutation screening therefore may be considered in athletes presenting with presenile onset of behavioral disturbances and parkinsonism. Likewise, as the frequency of presenilin-1 (PSEN1) mutation (the most common pathogenic mutation in AD) increases with early onset dementia and strength of family history,50, 80 autosomal dominant AD may be considered in middle-aged athletes presenting with dementia and possible family history. The occasional cases in the DP literature with middle age onset dementia and frank AD pathology,5 for example, suggest early onset AD rather than DP.5, 50 Finally, although only 10% of Parkinson disease patients have a positive family history, some pathogenic mutations produce substantia nigra degeneration without synucleinopathy (e.g., LRRK, parkin),81 which may be considered in athletes with early onset parkinsonism, substantia nigra degeneration at autopsy, and no Lewy bodies.
Neuropathology as a Predictor of Function in Degenerative Proteinopathies
The strict association between ptau lesions and behavioral or dysexecutive symptoms, and indeed the implied causal link between the two,18, 82 is problematic in that autopsy brain examination, including ptau immunohistochemistry, is useful for structural neuropathology. Neuropsychiatric signs, in contrast, are functional or biochemical. In point of fact, correlating basic neurologic dysfunction even with rigorously quantitated lesions and the availability of copious cognitive data is challenging.83 Blinded neuropathologic examination, for example, cannot distinguish intact cognition from dementia in the very old.84 Discerning clinical presentation among the FTD subtypes, or initial presentation in Lewy body diseases (cortical signs versus parkinsonism),74, 85, 86, 87 also cannot be carried out accurately with available methodology and consensus criteria. With this in mind, drawing mechanistic associations between tauopathy at autopsy, and psychiatric signs or complex behaviors such as suicide, explosive anger, impulse control, or posttraumatic stress disorder,88 appears beyond the scope of neuropathological interpretation.
Neurodegenerative Disease Risk with Athletic Exposure
In a recent study of former NFL players by Lehman et al,89 the mortality rate from neurodegenerative disease was three times greater than that of the general population, whereas the mortality rate overall of NFL players was about half that of the general population. The numbers in this study were relative small, with 334 former players, of which there were two cases of AD and six ALS cases. Moreover, none of these cases had autopsy confirmation or genetic screening. One study of professional Italian soccer players showed an increased risk of ALS,90 although selection bias and possible confounding influences of dietary and environmental factors have been raised.91 Physical activity in general has also been suggested as a risk for ALS,92 but an association with physical activity per se has been generally refuted.93 The only class II study of ALS incidence in American football found no risk.94 The improved mortality in the Lehman et al study is also interesting in that it raises the issue of health benefit of sport, which is only rarely discussed in the context of CTE.16 Regardless, the available evidence does not support a cause–effect relationship between exposure to head trauma in contact sports and neurodegenerative disease.
Arguably, the most substantial shortfall of the autopsy studies on DP and CTE, and autopsy studies in general, is the lack of prevalence data. The relationship between CTE changes at autopsy and neuropsychiatric signs and symptoms is therefore an open question, as is the extent and impact of susceptibility factors, kinetics of progression, and whether CTE exists as a distinct clinicopathologic entity. It is also noteworthy that NFL athletes do not appear to have an increased risk of suicide compared with the general population (data suggest a decreased risk),95, 96 casting some doubt on the inference that concussive or subconcussive exposure leads to heightened suicidality.
The Role of Neuroplasticity in Repetitive Head Trauma
Parents, athletes, and perhaps combat veterans may find solace in the fact that one can survive a career in the NFL neurologically intact and with no significant proteinopathy. Figure 2, for example, depicts the only brain finding identified in a retired, middle-aged NFL player. This individual came to autopsy after passing away from natural causes, with no history of neurologic or psychiatric illness. He further played on the offensive line, a position that suffers the most frontal impacts of all positions in the sport.97 He retired after several years in the NFL, having also played for 5 years at a major Division I university. A conservative estimate, based indirectly on accelerometer studies, is that he suffered as many as 10 000 head impacts exceeding 10 times g, over the course of just his collegiate and professional careers.98 This athlete again had no neurologic or psychiatric disturbances during life, and, at autopsy, his brain was entirely normal by gross and microscopic examination. The only incidental finding, after extensive examination of the brain for ptau throughout the neuraxis was a small collection of neurofibrillary tangles and neuropil threads in the superficial amygdala, and a pretangle and rare neuropil threads in the locus ceruleus. This extent of ptau may be found in any individual of this age, irrespective of APOE, occupation, head trauma exposure, or other factors.

Focal neurofibrillary degeneration in the superficial amygdala was noted in this 41-year-old retired NFL offensive lineman who, by definition, suffered thousands of head impacts over the course of this career (scale bar=400 μm). This was the only finding noted after an extensive examination for ptau (AT8) throughout the neuraxis, and is an incidental finding for age. This is the oldest NFL athlete reported to date with no CTE changes at autopsy.
Although this negative case may not appear noteworthy on casual review, it should be pointed that this is the first reported middle-aged NFL athlete to date with no CTE changes.1, 2 This raises the issue of the much needed prevalence data and selection biases, if not the resilience and plasticity of the human brain, and its ability to endure protracted physical punishment in the setting of high-energy collision sports at the highest level.
CONCLUSIONS
The recent CTE literature has advanced the discussion beyond the preceding CTE literature, by codifying a specific distribution of proteinopathy, and laying out criteria for future studies. The criteria for CTE nevertheless appear overly inclusive, which may in turn hamper understanding of molecular-genetic underpinnings. It may also be acknowledged that there are more questions than answers about all aspects of the CTE concept, from biomechanical substrates to molecular pathogenesis to the existence of CTE as a distinct entity.
References
McKee AC, Stern RA, Nowinski CJ et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013;136:43–64.
Omalu B, Bailes J, Hamilton RL et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery 2011;69:173–183.
Martland HS . Punch Drunk. JAMA 1928;91:1103–1107.
Millspaugh JA . Dementia Pugilistica. US Naval Med Bulletin 1937;35:297–361.
Corsellis JA, Bruton CJ, Freeman-Browne D . The aftermath of boxing. Psychol Med 1973;3:270–303.
Geddes JF, Vowles GH, Robinson SF et al. Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol 1996;22:12–16.
McCrory P, Meeuwisse WH, Kutcher JS et al. What is the evidence for chronic concussion-related changes in retired athletes: behavioural, pathological and clinical outcomes? Br J Sports Med 2013;47:327–330.
Gardner A, Iverson GL, McCrory P . Chronic traumatic encephalopathy in sport: a systematic review. Br J Sports Med 2014;48:84–90.
Roberts GW, Allsop D, Bruton C . The occult aftermath of boxing. J Neurol Neurosurg Psychiatry 1990;53:373–378.
Mawdsley C, Ferguson FR . Neurological disease in boxers. Lancet 1963;2:795–801.
Payne EE . Brains of boxers. Neurochirurgia (Stuttg) 1968;11:173–188.
Geddes JF, Vowles GH, Nicoll JA et al. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 1999;98:171–178.
Nowak LA, Smith GG, Reyes PF . Dementia in a retired world boxing champion: case report and literature review. Clin Neuropathol 2009;28:275–280.
Roberts AH . Brain damage in boxers: a study of the prevalence of traumatic encephalopathy among ex-professional boxers. Pitman: London, UK, 1969.
Bailes JE, Petraglia AL, Omalu BI et al. Role of subconcussion in repetitive mild traumatic brain injury. J Neurosurg 2013;119:1235–1245.
McKee AC, Daneshvar DH, Alvarez VE et al. The neuropathology of sport. Acta Neuropathol 2014;127:29–51.
Williams DJ, Tannenberg AE . Dementia pugilistica in an alcoholic achondroplastic dwarf. Pathology 1996;28:102–104.
Goldstein LE, Fisher AM, Tagge CA et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 2012;4:134ra160.
DeKosky ST, Blennow K, Ikonomovic MD et al. Acute and chronic traumatic encephalopathies: pathogenesis and biomarkers. Nat Rev Neurol 2013;9:192–200.
Rosenfeld JV, McFarlane AC, Bragge P et al. Blast-related traumatic brain injury. Lancet Neurol 2013;12:882–893.
Harmon KG, Drezner JA, Gammons M et al. American Medical Society for Sports Medicine position statement: concussion in sport. Br J Sports Med 2013;47:15–26.
Rosenthal JA, Foraker RE, Collins CL et al. National High School Athlete Concussion Rates From 2005-2006 to 2011-2012. Am J Sports Med 2014;42:1710–1715.
Harmon KG, Drezner J, Gammons M et al. American Medical Society for Sports Medicine position statement: concussion in sport. Clin J Sport Med 2013;23:1–18.
Cantu RC . Chronic traumatic encephalopathy in the National Football League. Neurosurgery 2007;61:223–225.
Blumbergs PC, Scott G, Manavis J et al. Staining of amyloid precursor protein to study axonal damage in mild head injury. Lancet 1994;344:1055–1056.
Dawson SL, Hirsch CS, Lucas FV et al. The contrecoup phenomenon. Reappraisal of a classic problem. Hum Pathol 1980;11:155–166.
Holbourne A . The mechanics of head injuries. British Medical Bulletin 1945;3:147–149.
Gennarelli TA, Thibault LE, Adams JH et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol 1982;12:564–574.
Adams JH, Graham DI, Gennarelli TA . Head injury in man and experimental animals: neuropathology. Acta Neurochir Suppl (Wien) 1983;32:15–30.
Gennarelli TA . Head injury in man and experimental animals: clinical aspects. Acta Neurochir Suppl (Wien) 1983;32:1–13.
Adams JH, Doyle D, Ford I et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 1989;15:49–59.
Morrison B 3rd, Elkin BS, Dolle JP et al. In vitro models of traumatic brain injury. Annu Rev Biomed Eng 2011;13:91–126.
Xiong Y, Mahmood A, Chopp M . Animal models of traumatic brain injury. Nat Rev Neurosci 2013;14:128–142.
Augustine C, Cepinskas G, Fraser DD et al. Traumatic injury elicits JNK-mediated human astrocyte retraction in vitro. Neuroscience 2014;274C:1–10.
Gatson JW, Warren V, Abdelfattah K et al. Detection of beta-amyloid oligomers as a predictor of neurological outcome after brain injury. J Neurosurg 2013;118:1336–1342.
Begum G, Yan HQ, Li L et al. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci 2014;34:3743–3755.
Cornelius C, Crupi R, Calabrese V et al. Traumatic brain injury: oxidative stress and neuroprotection. Antioxid Redox Signal 2013;19:836–853.
Balan IS, Saladino AJ, Aarabi B et al. Cellular alterations in human traumatic brain injury: changes in mitochondrial morphology reflect regional levels of injury severity. J Neurotrauma 2013;30:367–381.
Woo SK, Kwon MS, Ivanov A et al. The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J Biol Chem 2013;288:3655–3667.
Giunta B, Obregon D, Velisetty R et al. The immunology of traumatic brain injury: a prime target for Alzheimer's disease prevention. J Neuroinflammation 2012;9:185.
Wong J, Hoe NW, Zhiwei F et al. Apoptosis and traumatic brain injury. Neurocrit Care 2005;3:177–182.
Tran HT, Sanchez L, Brody DL . Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J Neuropathol Exp Neurol 2012;71:116–129.
Lucke-Wold BP, Turner RC, Logsdon AF et al. Linking traumatic brain injury to chronic traumatic encephalopathy: identification of potential mechanisms leading to neurofibrillary tangle development. J Neurotrauma 2014;31:1129–1138.
Mohamed NV, Herrou T, Plouffe V et al. Spreading of tau pathology in Alzheimer's disease by cell-to-cell transmission. Eur J Neurosci 2013;37:1939–1948.
de Calignon A, Polydoro M, Suarez-Calvet M et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 2012;73:685–697.
Clavaguera F, Akatsu H, Fraser G et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 2013;110:9535–9540.
Watts JC, Condello C, Stohr J et al. Serial propagation of distinct strains of Abeta prions from Alzheimer's disease patients. Proc Natl Acad Sci USA 2014;111:10323–10328.
Stohr J, Condello C, Watts JC et al. Distinct synthetic Abeta prion strains producing different amyloid deposits in bigenic mice. Proc Natl Acad Sci USA 2014;111:10329–10334.
Peskind ER, Brody D, Cernak I et al. Military- and sports-related mild traumatic brain injury: clinical presentation, management, and long-term consequences. J Clin Psychiatry 2013;74:180–188.
Brandenburg W, Hallervorden J . Dementia pugilistica with anatomical findings. Virchows Arch 1954;325:680–709.
Courville CB . Punch drunk. Its pathogenesis and pathology on the basis of a verified case. Bull Los Angel Neuro Soc 1962;27:160–168.
Constantinides J, Tissot R . Lesions neurofibrillaires d'Alzheimer generalisees sans plaques seniles. Archives Suisses de Neurologie, Neurochirurgie et de Psychiatrie 1967;100:117–130.
Clausen H, McCrory P, Anderson V . The risk of chronic traumatic brain injury in professional boxing: change in exposure variables over the past century. Br J Sports Med 2005;39:661–664.
Tokuda T, Ikeda S, Yanagisawa N et al. Re-examination of ex-boxers' brains using immunohistochemistry with antibodies to amyloid beta-protein and tau protein. Acta Neuropathol 1991;82:280–285.
Hof PR, Knabe R, Bovier P et al. Neuropathological observations in a case of autism presenting with self-injury behavior. Acta Neuropathol 1991;82:321–326.
Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 12-1999. A 67-year-old man with three years of dementia. N Engl J Med 1999;340:1269–1277.
Schmidt ML, Zhukareva V, Newell KL et al. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer's disease. Acta Neuropathol 2001;101:518–524.
Areza-Fegyveres R, Rosemberg S, Castro RM et al. Dementia pugilistica with clinical features of Alzheimer's disease. Arq Neuropsiquiatr 2007;65:830–833.
Hardy JA, Higgins GA . Alzheimer's disease: the amyloid cascade hypothesis. Science 1992;256:184–185.
Castellani RJ, Nunomura A, Lee HG et al. Phosphorylated tau: toxic, protective, or none of the above. J Alzheimers Dis 2008;14:377–383.
Smith MA, Taneda S, Richey PL et al. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci USA 1994;91:5710–5714.
Sayre LM, Zelasko DA, Harris PL et al. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem 1997;68:2092–2097.
Sayre LM, Perry G, Harris PL et al. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals. J Neurochem 2000;74:270–279.
Gomez-Isla T, Hollister R, West H et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 1997;41:17–24.
Santacruz K, Lewis J, Spires T et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005;309:476–481.
Small GW, Kepe V, Siddarth P et al. PET scanning of brain tau in retired national football league players: preliminary findings. Am J Geriatr Psychiatry 2013;21:138–144.
Crary JF, Trojanowski JQ, Schneider JA et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–766.
Braak H, Del Tredici K . The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol 2011;121:171–181.
Braak H, Thal DR, Ghebremedhin E et al. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011;70:960–969.
Liu L, Drouet V, Wu JW et al. Trans-synaptic spread of tau pathology in vivo. PLoS One 2012;7:e31302.
Lewis J, Dickson DW, Lin WL et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001;293:1487–1491.
Jones BE . Noradrenergic locus coeruleus neurons: their distant connections and their relationship to neighboring (including cholinergic and GABAergic) neurons of the central gray and reticular formation. Prog Brain Res 1991;88:15–30.
Jordan BD, Relkin NR, Ravdin LD et al. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. JAMA 1997;278:136–140.
Rademakers R, Neumann M, Mackenzie IR . Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 2012;8:423–434.
Renton AE, Chio A, Traynor BJ . State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014;17:17–23.
Bennion Callister J, Pickering-Brown SM . Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS. Exp Neurol 2014;262:84–90.
Liu Y, Yu JT, Sun FR et al. The clinical and pathological phenotypes of frontotemporal dementia with C9ORF72 mutations. J Neurol Sci 2013;335:26–35.
Keage HA, Hunter S, Matthews FE et al. TDP-43 in the population: prevalence and associations with dementia and age. J Alzheimers Dis 2014;42:641–650.
Mackenzie IR, Neumann M, Bigio EH et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 2010;119:1–4.
Loy CT, Schofield PR, Turner AM et al. Genetics of dementia. Lancet 2014;383:828–840.
Klein C, Westenberger A . Genetics of Parkinson's disease. Cold Spring Harb Perspect Med 2012;2:a008888.
Omalu B, Hammers JL, Bailes J et al. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus 2011;31:E3.
Castellani RJ, Lee HG, Zhu X et al. Alzheimer disease pathology as a host response. J Neuropathol Exp Neurol 2008;67:523–531.
Brayne C, Richardson K, Matthews FE et al. Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge city over-75s cohort (CC75C) Study. J Alzheimers Dis 2009;18:645–658.
Llado A, Sanchez-Valle R, Rey MJ et al. Clinicopathological and genetic correlates of frontotemporal lobar degeneration and corticobasal degeneration. J Neurol 2008;255:488–494.
Goldman JG, Williams-Gray C, Barker RA et al. The spectrum of cognitive impairment in Lewy body diseases. Mov Disord 2014;29:608–621.
Lehmann M, Ghosh PM, Madison C et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer's disease. Brain 2013;136:844–858.
McKee AC, Robinson ME . Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement 2014;10:S242–S253.
Lehman EJ, Hein MJ, Baron SL et al. Neurodegenerative causes of death among retired National Football League players. Neurology 2012;79:1970–1974.
Chio A, Benzi G, Dossena M et al. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain 2005;128:472–476.
Beghi E . Are professional soccer players at higher risk for ALS? Amyotroph Lateral Scler Frontotemporal Degener 2013;14:501–506.
Huisman MH, Seelen M, de Jong SW et al. Lifetime physical activity and the risk of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2013;84:976–981.
Hamidou B, Couratier P, Besancon C et al. Epidemiological evidence that physical activity is not a risk factor for ALS. Eur J Epidemiol 2014;29:459–475.
Savica R, Parisi JE, Wold LE et al. High school football and risk of neurodegeneration: a community-based study. Mayo Clin Proc 2012;87:335–340.
Baron SL, Hein MJ, Lehman E et al. Body mass index, playing position, race, and the cardiovascular mortality of retired professional football players. Am J Cardiol 2012;109:889–896.
Iverson GL . Chronic traumatic encephalopathy and risk of suicide in former athletes. Br J Sports Med 2014;48:162–174.
Crisco JJ, Wilcox BJ, Beckwith JG et al. Head impact exposure in collegiate football players. J Biomech 2011;44:2673–2678.
Crisco JJ, Fiore R, Beckwith JG et al. Frequency and location of head impact exposures in individual collegiate football players. J Athl Train 2010;45:549–559.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest. The contents are solely the responsibility of the author and any opinions expressed herein are not necessarily the views of the editors, the United States and Canadian Academy of Pathology, or Nature Publishing Group.
Additional information
This editorial review highlights the evolving paradigm surrounding chronic traumatic encephalopathy (CTE), and gaps in knowledge which require further study. An in-depth understanding of CTE by the pathology community is critical to scientific investigation, as current concepts surrounding this entity have broad implications in society, from athletic participation to armed conflict.
Rights and permissions
About this article

Cite this article
Castellani, R. Chronic traumatic encephalopathy: A paradigm in search of evidence?. Lab Invest 95, 576–584 (2015). https://doi.org/10.1038/labinvest.2015.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2015.54
