
Abstract
Tumor necrosis factor-α (TNF-α) acts as a key factor for the development of inflammatory bowel diseases (IBDs), whose function is known to be mediated by TNF receptor 1 (TNFR1) or TNFR2. However, the precise role of the two receptors in IBD remains poorly understood. Herein, chronic colitis was established by oral administration of dextran sulfate sodium (DSS) in TNFR1 or TNFR2−/− mice. Unexpectedly, TNFR1 or TNFR2 deficiency led to exacerbation of signs of colitis compared with wild-type (WT) counterparts. Of note, TNFR1 ablation rendered significantly increased mortality compared with TNFR2 and WT mice after DSS. Aggravated pathology of colitis in TNFR1−/− or TNFR2−/− mice correlated with elevated colonic expression of proinflammatory cytokines and chemokines. Importantly, ablation of TNFR1 or TNFR2 increased apoptosis of colonic epithelial cells, which might be due to the heightened ratio of Bax/Bcl-2 and increased expression of caspase-8. Intriguingly, despite comparable intensity of intestinal inflammation in TNFR-deficient mice after DSS, systemic inflammatory response (including splenomegaly and myeloid expansion) was augmented dramatically in TNFR1−/− mice, instead of TNFR2−/− mice. Granulocyte-macrophage colony-stimulating factor (GMCSF) was identified as a key mediator in this process, as neutralization of GMCSF dampened peripheral inflammatory reaction and reduced mortality in TNFR1−/− mice. These data suggest that signaling via TNFR1 or TNFR2 has a protective role in chronic intestinal inflammation, and that lacking TNFR1 augments systemic inflammatory response in GMCSF-dependent manner.
Similar content being viewed by others


Main
Tumor necrosis factor (TNF) has a crucial role in immune response under physiological and pathological conditions, which is largely ascribed to its pleiotropic function on differentiation, growth and apoptosis of both immune and non-immune cells.1, 2 It is well established that TNF is tightly implicated in the pathogenesis of inflammatory diseases and autoimmunity such as rheumatoid arthritis, multiple sclerosis and inflammatory bowel diseases (IBDs).3 TNF exerts its functions by ligation of either TNF receptor (TNFR) 1 or TNFR2 as a trimer. TNFR1, which is expressed on most cell types, contains a death domain that delivers apoptosis signaling through caspase activation and promotes nuclear translocation of nuclear factor (NF)-κB, resulting in transcription of inflammatory cytokines, chemokines and anti-apoptotic molecules.4 TNFR2, which is mainly expressed by hematopoietic cell populations, lacks the death domain but is able to mediate apoptosis through the kinase receptor-interacting protein.5
Currently, accumulating evidence clearly supports the intimate relationship of TNFR expression with the pathogenesis of IBD. During active stages of the disease, TNFR2 expression on colonic epithelial cells is elevated drastically in patients with IBD and in rodents with experimental colitis.6, 7 Moreover, polymorphisms in the TNFR2 gene have been reported to be associated with a higher susceptibility to Crohn’ disease.8 These demonstrations imply a disease-promoting role of TNF signaling via TNFR2 during IBD development. However, a recent study delineates an opposite effect of TNFR2 on the course of T-cell-mediated colitis, that the transfer of colitogenic CD4+CD45hiTNFR2−/− T cells into RAG−/− recipients leads to acceleration of the onset of overt disease and to aggravated severity of intestinal inflammation.9 The paradoxical roles of TNF signaling via TNFR1 in CD also exist. Ebach et al10 reported that TNFR1−/− C57Bl/6 mice had more weight loss and increased mortality after trinitrobenzene sulphonic acid (TNBS) instillation. In contrast, Nakai et al11 demonstrated that TNFR1 ablation attenuated tissue damage after TNBS, which was related to reduced NF-κB activity. Our recent study has shown that TNF signaling via TNFR1 or TNFR2 have a pathogenic role in TNBS-induced colitis.12 Overall, these studies underscore the complexity of TNF signaling via TNFR1 or TNFR2 during the onset and perpetuation of intestinal inflammation, which may be affected by different TNFR expression patterns and distinct colitis models used.
The studies on the role of TNF signaling via TNFR1 or TNFR2 in the course of dextran sulfate sodium (DSS)-induced colitis, which is another widely used murine model of IBD and closely resembles ulcerative colitis (UC), is relatively few. Recently, we have demonstrated that two receptors (TNFR1 and TNFR2) have distinct roles during acute intestinal inflammation. TNFR1−/− Balb/c mice displayed more severe mucosal damage, whereas TNFR2 knockout led to attenuated pathology of colitis when acute intestinal inflammation was induced by DSS instillation.13 However, the precise role of TNF signaling via TNFR1 or TNFR2 during the initiation and progression of chronic intestinal inflammation remains to be elucidated.
In this study, chronic colitis in TNFR1−/−, TNFR2−/− and wild-type (WT) mice was established by sequential ingestion of DSS in distilled water. We found that TNF signaling via TNFR1 or TNFR2 had a protective role in the course of chronic intestinal inflammation. It is noteworthy that TNFR1 deficiency rendered more mortality compared with TNFR2−/− and WT mice, which may be due to augmented systemic inflammatory response in TNFR1−/− mice after DSS. Furthermore, granulocyte macrophage colony-stimulating factor (GMCSF) was identified as a key player in mounting lethal inflammatory response, as in vivo neutralization of GMCSF dampened peripheral inflammatory reaction and reduced mortality in TNFR1−/− mice.
MATERIALS AND METHODS
Animals
BALB/c WT mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). TNFR1−/− and TNFR2−/− mice from Dr Zhihai Qin (National Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing, China) were backcrossed for 12 generations onto the BALB/c background.14 Animals were housed in specific pathogen-free conditions with an alternating light–dark cycle. All experiments were performed using 6- to 8-week-old male mice. Care, use and treatment of mice in this study were in strict agreement with international guidelines for the care and use of laboratory animals and approved by Animal Ethics Committee of Institute of Basic Medical Sciences.
Chronic Colitis Induction
Mice received water with 2% DSS (MW 36 000–50 000; MP Biochemicals, OH, USA) for 7 days. After this, mice were kept on regular water for 14 days and subjected to three more DSS treatment cycles. Weights were measured before induction of colitis and daily thereafter. On day 78, mice were killed by cervical dislocation under anesthesia.
Histology
Colons or spleen were removed and fixed in 10% neutral-buffered formalin solution and then embedded in paraffin, cun into tissue sections, and stained with hematoxylin and eosin. Stained sections were examined for evidence of colitis using a criteria described previously:15 the presence of lymphocyte infiltration, elongation and/or distortion of crypts, frank ulceration, and thickening of the bowel wall. The degree of inflammation on microscopic cross-sections of the colon was graded semiquatitatively from 0 to 4 (0, no evidence of inflammation; 1, low level of lymphocyte infiltration with infiltration seen in <10% high-power field (hpf), no structural changes observed; 2, moderate lymphocyte infiltration with infiltration seen in 10–25% hpf, crypt elongation, bowel wall thickening that does not extend beyond mucosal layer, no evidence of ulceration; 3, high level of lymphocyte infiltration with infiltration seen in 25–50% hpf, high vascular density, thickening of bowel wall that extends beyond mucosal layer; and 4, marked degree of lymphocyte infiltration with infiltration seen in >50% hpf, high vascular density, crypt elongation with distortion, transmural bowel wall thickening with ulceration).
Isolation of Colonic Epithelial Cells
Colonic epithelial cells were isolated according to the protocol proposed previously,15 with a mild modification. Briefly, the entire length of colon was opened longitudinally, washed with PBS containing 100 μg/ml gentamycin, and immersed in fresh ice-cold PBS supplemented with protease inhibitors and phosphate-inhibitors (0.2 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 1 mM benzamidine, 1 mM sodium orthovanadate and 2 μM cantharidin). The bowel was then cut into small pieces and put into 5 ml of ice-cold BSS buffer (1.5 mM KCl, 96 mM NaCl, 27 mM sodium citrate, 8 mM KH2PO4, 5.6 mM Na2HPO4, 15 mM EDTA and 1 mM dithiothreitol) with inhibitors as above, vortexed at 4 °C at maximum speed for 5 min. This process was repeated two additional times for 3 and 7 min, respectively. After a total of 15 min of vortexing, almost all epithelial cells were recovered in the BSS solution. The solution was then spun down at 1000 g for 10 min at 4 °C and washed once with Tris buffer (150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 50 mM Tris-HCl, pH 8.0) with inhibitors as above. The resulting pellets were collected as colonic epithelial cells (purity>80%, identified by CK-18 staining) and saved at −80 °C for protein experiments.
Detection of Apoptotic Cells
For detection of apoptotic cells in paraffin-embedded tissues, labeling of degraded DNA specific to apoptotic cells was performed using a modification of the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) technique by application of the in situ cell death detection kits (Roche Diagnostics, IN, USA) according to the manufacturer’s instructions. Levels of apoptosis were quantified by counting the number of TUNEL-positive cells per 100 nuclei. The apoptotic index was obtained from the ratio of apoptotic to total cells.
Colon Homogenates
A 1 cm segment was divided from the distal 4 cm of the harvest colon. Wet weight was recorded separately for the whole distal 4 cm and the portion taken for homogenation. Colon tissue samples were homogenized in PBS containing a cocktail of protease inhibitors (1 μl–20 mg of tissue according to the manufacturer’s protocol, Roche Diagnostics) with a Polytron homogenizer and centrifuged at 12 000 g for 10 min. The supernatants were stored at −20 °C until used for ELISA analysis.
Measurement for Colon Edema
Colons were dissected at indicated time point after TNBS. A piece of the affected colon was then collected, weighed, placed in an 80 °C oven for 24 h, then reweighed, and the wet-to-dry weight ratio was determined as a measure of colon edema.16
Measurement of Myeloperoxidase Activity
Tissue MPO activity was determined by a standard enzymatic procedure as described previously,17 with minor modifications. Briefly, after the samples were weighed, a tissue sample (approximately 300 mg) was homogenized in a buffer (0.5% hexadecyltrimethylammonium bromide in 50 mM potassium phosphate buffer, pH 6.0) three times for 30 s each on ice. The sample was centrifuged at 20 000 g for 20 min at 4 °C and the supernatant was collected. The supernatants (100 μl) was then added to 2.9 ml of 50 mM phosphate buffer (pH 6.0) containing 0.167 mg/ml O-dianisidine hydrochloride and 0.0005% hydrogen peroxide, and absorbances were measured using a spectrometer at 25 °C. Results for colon MPO content were converted to absorbance units per gram of tissue.
Immunoblotting
Samples were homogenized and sonicated in RIPA lysis buffer (Santa Cruz Biotechnology, TX, USA), supplemented with protease inhibitors. After centrifugation at 20 000 g for 15 min, 30 μg of the supernatants were separated on 10% SDS-polyacrylamide gel and transferred onto an Immunobilon-P Transfer membrane (Millipore, MA, USA). After being blocked with 5% skim milk, the membrane was incubated with antibodies to Bcl-2 (1:1000), Bcl-xL (1:1000), Bax (1:1000), Bad (1:1000), caspase 8 (1:1000) or caspase 3/cleaved caspase 3 (1:1000). Rabbit anti-actin antibody (1:1000) was used as an internal control. ImmunoPure peroxidase-conjugated anti-rabbit IgG were used as secondary antibodies. The blotted membrane was then treated with the Super Signal West Dura Extended Duration Substrate (Thermo, IL, USA) and signals were detected by LAS-3000 mini CCD camera (Fuji Film, Tokyo, Japan). Primary antibodies used for immunoblotting were purchased from eBioscience (CA, USA), except for antibody to actin (Sungene, Tianjin, China).
Cytokine Analysis
Colonic interleukin (IL)-6, IL-1β, monocyte chemotactic protein (MCP)-1, IL-17A and serum GMCSF, GCSF, MCSF, IL-3 were measured by ELISA according to the manufacturer’s instructions. ELISA kits were purchased from eBioscience.
Flow Cytometry
Single-cell suspension was prepared from spleen or bone marrow by passing them through nylon mesh and stained with appropriate antibodies in PBS with 2% heat-inactivated fetal calf serum and 0.1% sodium azide on ice for 25 min. The antibodies used are following: fluorescein isothiocyanate (FITC)-labeled anti-murine Gr-1 (RB6-8C5, Biolegend, CA, USA), F4/80 (CI:A3-1, Biolegend) antibody and corresponding isotypes (Biolegend). These cells were fixed using PBS with 1% paraformaldehyde. Data collection and analysis were performed on a FACS Calibur flow cytometer using CellQuest software (Becton Dickinson, NJ, USA).
In Vivo Neutralization
Mice subjected to induction of chronic colitis received intraperitoneally monoclonal antibody to GMCSF (MP1-22E9, Biolegend) or IL-3 (MP2-8F8) at a dose of 0.25 mg/mouse on days −1, 0, 2, 5 and then every 5 days until animals were killed. As controls, rat IgG was used at the same dose and periodicity.
Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
RNA extracted from colon tissues was reverse transcribed into cDNA using the SuperScript III First Strand cDNA synthesis system (Invitrogen, CA, USA). cDNA was synthesized from 0.5 μg RNA using random hexamer primers and SuperScript III(Invitrogen). Real-time RT-PCR was performed on a Bio-Rad iCycler to quantify mRNA levels. The primers for real-time were as follows: CXCL-1 forward primer 5′-ACTGCACCCAAACCGAAGTC-3′ and reverse primer 5′-TGGGGACACCTTTTAGCATCTT-3′; CXCL-2 forward primer 5′-GCGCCCAGACAGAAGTCATAG-3′ and reverse primer 5′-AGCCTTGCCTTTGTTCAGTATC-3′; CCL-3 forward primer 5′-TGTACCATGACACTCTGCAAC-3′ and reverse primer 5′-CAACGATGAATTGGCGTGGAA-3′; GAPDH forward primer 5′-TGAAGGTCGGAGTCAACGGATTTGGT-3′ and reverse primer 5′-CATGTGGGCCATGAGGTCCACCAC-3′. All reactions were performed in triplicate. The data were analyzed using Q-Gene software and expressed as fold change mean normalized expression (MNE) from control value. MNE is directly proportional to the amount of RNA of the target gene relative to the amount of RNA of the reference gene, GAPDH.
Student’s t-test and one-way analysis of variance was used to determine significance, with P<0.05 considered significant. Kaplan–Meier was used for survival analysis with log-rank P<0.05 to determine significance. Statistics were performed using SPSS 10.0 for Macintosh, and graphs were made on Deltagraph (SPSS, Chicago, IL, USA).
RESULTS
TNF Signaling Via TNFR1 or TNFR2 Confers Protection Against Weight Loss and Mortality
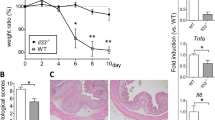
Chronic colitis was established in WT and TNFR-knockout mice by four-cycle drinking of DSS (2%) for 78 days, respectively. In contrast to those with acute colitis, which exhibited profound weight loss,13 WT mice suffering from chronic colitis had no significant loss of body weight, compared with WT controls (Figure 1a). TNFR1−/− and TNFR2−/− mice, however, harbored 60.74±7.92% and 60.87±9.61% of the weight of WT counterparts, respectively, on day 78 after initiation of drinking DSS (Figure 1a). No significant difference was observed between TNFR1−/− and TNFR2−/− groups. Interestingly, at the end of experiments (on day 78 after DSS), 59% (19/32) of TNFR1−/−, and 92% (22/24) of TNFR2−/− mice had survived, whereas survival was 100% (22/22) in WT mice (Figure 1b).

TNFR1 or TNFR2−/− mice have more weight loss and mortality after DSS. Chronic DSS colitis was established as described in Materials and methods section. (a) Weight in each group was monitored daily at 78-day intervals. Weight was presented as percentage of the initial weight at day 0. (b) Survival rate was calculated at the end of experiments. Data are pooled from three independent experiments (number in each group was indicated).
Absence of TNFR1 OR TNFR2 Exacerbates Intestinal Damage after DSS
Next, we examined the influence of TNFR1 or TNFR2 deficiency on macroscopic and microscopic injury of colons after DSS. WT mice displayed significant shortening of the colons after DSS (Figures 2a and b). In accord with the change of body weight, TNFR1−/− and TNFR2−/− mice showed more considerable injury (Figures 2a and b). Furthermore, wet-to-dry weight ratios of colons from TNFR1−/− and TNFR2−/− mice were remarkably increased compared with DSS-instilled WT littermates (Figure 2c). Consistent with macroscopic alterations, WT mice after DSS showed numerous infiltration of inflammatory leukocytes and ulcerative injury. TNFR1 or TNFR2 ablation rendered more severity of histopathological features of colitis (Figure 2d). When quantified by histological system for evidence of inflammation and injury, these histological alterations were highly significant (Figure 2e).

TNFR1 or TNFR2 deficiency exacerbates mucosal damage after DSS. Colons from each group were collected on day 78 after DSS instillation. (a) Macroscopic images of the colons are shown. (b) The length of colon was measured. (c) Colon edema was determined by wet-to-dry weight ratios. (d) Microscopic images of the colons are shown by hematoxylin and eosin staining. Magnification × 200. (e) Colitis score was determined as described in Materials and methods section. Data represent the mean±s.d. of 8–10 mice per group. **P<0.01 vs WT control mice; #P<0.05, ##P<0.01 vs WT mice after DSS.
Mice Lacking TNFR1 or TNFR2 have Higher Levels of MPO in Colons after DSS
Neutrophil infiltrate is a key event for initiation and promotion of inflammatory responses in many inflammatory diseases including IBD.18, 19, 20 To determine whether severe signs of colitis in mice lacking TNFR1 or TNFR2 are partially because of increased infiltrate of neutrophil, we examined the levels of MPO, indices for neutrophil infiltration, in the colons on day 78 after DSS. The contents of MPO in the colons of WT colitis mice were much more than control mice (Figure 3). In mice lacking TNFR1 or TNFR2, the levels of colonic MPO were increased remarkably compared with WT mice after DSS (Figure 3).

TNFR1 or TNFR2−/− mice have increased infiltrates of neutrophils into the colons after DSS. Levels of myeloperoxidase (MPO) in the colons of mice in each group were measured on day 78 after colitis induction. Data represent the mean±s.d. of 4–5 mice per group. **P<0.01 vs WT control mice; #P<0.05 vs WT mice after DSS.
TNFR1 or TNFR2 Ablation Upregulates the Colonic Production of Proinflammatory Cytokines and Chemokines after DSS
To investigate whether disease-promoting effects of TNFR1 or TNFR2 deficiency after DSS is linked to the alteration of expression of colitis-related proinflammatory cytokines and chemokines, the section of colons was dissected, homogenized and assayed on day 78 after DSS. Compared with WT colitis mice, lack of TNFR1 or TNFR2 led to a significant increase of IL-6, IL-1β, MCP-1, IL-17A production in the colon tissues (Figure 4a). These results were in agreement with the data of colitis scores. Furthermore, the colonic expression of neutrophil-chemoattractant chemokines CXCL-1, CXCL-2 and CCL-3 was elevated in TNFR1−/− and TNFR2−/− mice, compared with WT counterparts (Figure 4b). This may explain the observation of the widespread infiltration of neutrophil in target tissues of TNFR1−/− and TNFR2−/− mice.

Lack of TNFR1 or TNFR2 increases proinflammatory mediator production in the colons after DSS. Colons were isolated and homogenized on day 78 after colitis induction. (a) IL-6, IL-1β, MCP-1 and IL-17A concentrations in the supernatants of homogenates were determined by ELISA. (b) mRNA was isolated from homogenates and CXCL1/2, CCL3 expression was examined by quantitative RT-PCR. Data represent the mean±s.d. of 6–8 mice per group from two independent experiments. **P<0.05 vs WT control mice; #P<0.05 vs WT mice after DSS.
TNFR1 or TNFR2 Deficiency Enhances the Activity of Apoptosis-Promoting Proteins in Colonic Epithelial Cells after DSS
To determine the mechanisms underlying aggravated intestinal damage in absence of TNFR1 or TNFR2 when chronic colitis was established, we hypothesize that lacking TNF signaling via TNFR1 or TNFR2 leads to increased apoptosis of colonic epithelial cells, thereby to more severity of colitis, considering that TNF is a key mediator for epithelial cell death and mucosal injury during inflammation.21, 22 To address this issue, colon samples from WT and TNFR-knockout groups were stained using the TUNEL methods. In agreement with the grade of mucosal damage, TNFR1−/− and TNFR2−/− mice, which had higher colitis scores, exhibited significant increase of apoptotic epithelial cells after DSS (Figure 5a).

TNFR1 or TNFR2 ablation increases apoptosis of colonic epithelial cells after DSS. (a) Colons were dissected on day 78 after colitis induction. Apoptosis was determined by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) staining. Quantification of apoptosis was shown as apoptotic index (see Materials and methods section). Data represent the mean±s.d. of 8–10 mice per group. (b) At the same time points, colonic epithelial were isolated and the proteins were extracted. Bax, Bcl-2, Bcl-xL, Bad, caspase-8, caspase-3 and cleaved caspase-3 expression was examined by western blotting. β-Actin was used as internal control. Changes in quantity of expression of these factors were determined by densitometric assays. Data are representative of three independent experiments. *P<0.05, **P<0.01 vs WT control mice; #P<0.05 vs WT mice after DSS.
Next, we investigate whether increase in apoptosis of colonic epithelial cells in TNFR-deficient mice after DSS is associated with enhanced pro-apoptosis signals. The results showed that the expression of pro-apoptotic factor Bax in epithelial cells of TNFR1−/− and TNFR2−/− mice was augmented dramatically compared with WT littermates after DSS (Figure 5b). Furthermore, Bcl-2 and Bcl-xL expression, two members of Bcl-2 family with anti-apoptotic function, was reduced in the epithelium of TNFR1−/− and TNFR2−/− groups (Figure 5b). Densitometry analysis further showed that the ratio of Bax/Bcl-2 and Bax/Bcl-xL in TNFR−/− mice was significantly higher than in WT mice after DSS (Figure 5b), which might account for enhanced apoptosis of colonic epithelial cells in TNFR1−/− and TNFR2−/− mice after DSS. We also detected Bad expression and found no significant alteration of Bad expression in epithelial cells between TNFR1−/−, TNFR2−/− and WT mice after DSS (Figure 5b), indicating that this factor might be not involved in apoptosis of colonic epithelial cells during intestinal inflammation. Moreover, as caspase-8 is reported to be a key factor in the turnover of colonic epithelium under steady-state and inflammatory conditions,23, 24, 25 we detected its expression in the epithelial cells after DSS. The colonic epithelial cells of TNFR1−/− and TNFR2−/− colitis mice contained more amounts of caspase-8 protein compared with WT counterparts (Figure 5b). Furthermore, cleavage of caspase-3 in colonic epithelial cells, another member of caspase family, was determined. Active caspase-3 was found in epithelial cells of DSS-instilled WT mice (Figure 5b). Importantly, cleavage of caspase-3 was more pronounced in epithelium of TNFR1−/− or TNFR2−/− mice than those of WT mice after DSS (Figure 5b). Thus, epithelial cell-specific enhanced expression of pro-apoptotic proteins may account for exacerbated signs of colitis after DSS.
Ablation of TNFR1 Augments Systemic Inflammatory Responses
It is accepted that systemic inflammatory response is hallmark of chronic colitis.26, 27 Therefore, we investigated the influence of TNFR1 or TNFR2 deficiency on the disease parameters. Indeed, WT mice displayed visible splenomegaly in terms of spleen weights after DSS. Similar effects were observed in TNFR2−/− DSS-instilled mice, but no significant difference was found between WT and TNFR2−/− counterparts (Figure 6a). Strikingly, TNFR1−/− mice exhibited more significant splenic enlargement compared with WT and TNFR2−/− groups after DSS (Figure 6a). In accord with this, widespread myelopoiesis was found in all groups with colitis, which was more significant in TNFR1−/− DSS-instilled mice (Figure 6b). These data indicate a crucial role of TNF signaling via TNFR1 for restraining DSS-induced systemic inflammatory reaction.

TNFR1 deficiency augments systemic inflammatory response after DSS. (a) On day 78 after colitis induction, spleen was isolated and imaged. The weight of spleen was examined. (b) At the same time point, histological examination of spleen was performed. The representative images from three independent experiments were shown. Scale bar: 200 μm. (c–f) The percentages of granulocytes (Gr-1+) (c, e) or monocytes (F4/80+) (d, f) in the spleens (c, d) and bone marrow (e, f) were examined by flow cytometry. Representative plots were shown. Data represent the mean±s.d. of 4–6 mice per group from three independent experiments. **P<0.01 vs WT control mice; #P<0.05, ##P<0.01 vs WT mice after DSS; $P<0.05, $$P<0.01 vs TNFR1−/− mice after DSS.
To define the characteristics of this systemic inflammatory response induced by DSS administration, FACS analysis for frequency of granulocytes and monocytes in spleen was performed. The percentage of granulocytes (Gr-1+) was increased dramatically in all DSS-instilled groups (Figure 6c). This increase was more pronounced in TNFR1−/− mice compared with WT and TNFR2−/− mice (Figure 6c). Similar effects were found in monocytes (F4/80+) expansion in the spleen (Figure 6d). We also detected the frequency of these cell populations in the bone marrow. The result showed that the percentage of granulocytes (Gr-1+) and monocytes (F4/80+) was increased dramatically in all DSS-instilled groups (Figures 6e and f). This increase was more pronounced in TNFR1−/− mice compared with WT and TNFR2−/− mice (Figures 6e and f), which was similar to the alteration in splenic compartments. Taken together, these data indicate that TNF signaling via TNFR1 is required for preventing hyper-responsive systemic inflammation, while TNFR2-mediated signaling does not.
GMCSF is Involved in Mediating Systemic Inflammatory Response in Absence of TNFR1 after DSS
To determine which factor is responsible for initiating systemic reaction in TNFR1−/−mice, we speculate that GMCSF, an important mediator for myelopoiesis,28 may be involved in this process. The results showed that the amounts of GMCSF in the serum of TNFR1−/− mice were much more than that of WT and TNFR2−/− mice after DSS (Figure 7a). It is noteworthy that under steady state, the contents of this cytokine were comparable in all groups (Figure 7a), suggesting that TNF signaling via TNFR1 or TNFR2 did not affect baseline release of GMCSF. Also, we detected G-CSF, M-CSF and IL-3 contents in the serum of WT, TNFR1−/−, TNFR2−/− mice with or without colitis. The data showed that all cytokine levels were increased in WT mice after DSS compared with naive littermates (Figure 7a). However, their contents in TNFR1−/− or TNFR2−/− DSS-instilled mice were comparable to WT counterparts (Figure 7a). Thus, TNFR1 or TNFR2 deficiency did not increase or decrease G-CSF, M-CSF and IL-3 contents during chronic intestinal inflammation and GMCSF release was selectively elevated in TNFR1−/− mice after DSS. Furthermore, blockade of GMCSF using neutralizing antibodies in vivo profoundly blunted systemic inflammatory response, as indicated by shrinking of spleen weight (Figure 7b), clearly supporting a key role of GMCSF in directing overt systemic inflammation because of lack of TNFR1 during chronic colitis. More importantly, blocking GMCSF in TNFR1−/− colitis mice dramatically reduced mortality (Figure 7c), indicating that GMCSF-mediated systemic inflammatory responses in TNFR1−/− mice causes less survival. Furthermore, to assess the effect of IL-3 on survival of TNFR1−/− DSS-instilled mice, neutralizing anti-IL-3 antibody was used. The result showed that blocking IL-3 bioactivity did not influence the survival rate of TNFR1−/− mice after DSS (Figure 7c). Thus, GMCSF, rather than IL-3, acts as a key player in mediating systemic inflammatory responses and increased mortality in TNFR1−/− mice after DSS.

Elevated production of GMCSF is required for augmented systemic inflammatory response and more mortality in TNFR1−/− mice after DSS. (a) On day 78 after colitis induction, serum was collected and GMCSF, GCSF, MCSF, IL-3 contents were examined by ELISA. Data represent the mean±s.d. of 5–10 mice per group from three independent experiments. *P<0.05 vs WT control mice; ##P<0.01 vs WT mice after DSS; $$P<0.01 vs TNFR1−/− mice after DSS. (b, c) WT, TNFR1−/− and TNFR2−/− mice were injected with anti-GMCSF or IL-3 antibody as described in Materials and methods section, followed by colitis induction. (b) Spleens were isolated and weighed. Data represent the mean±s.d. of 6–8 mice per group from two independent experiments. **P<0.01 vs TNFR1−/− mice receiving isotypes. (c) Survival rate in TNFR1−/− mice was calculated at the end of experiments. Data are pooled from two independent experiments (number in each group was indicated).
One question is raised whether neutralization of GMCSF also dampens local intestinal inflammation in TNFR1−/− mice, which may be a reason for increased survival in anti-GMCSF-treated groups. To address this, histopathological examination of colon tissues was performed. The results showed that blocking GMCSF bioactivity had no effect on mucosal injury in TNFR1−/− colitis mice (Figure 8a). The quantified histological evaluation also supported this conclusion (Figure 8b). Overall, these data indicate that GMCSF is a key factor involving in lethal systemic inflammatory reaction in absence of TNFR1.

Neutralizing GMCSF in TNFR1−/− mice has no effect on intestinal injury after DSS. TNFR1−/− mice were injected with anti-GMCSF antibody or isotypes as described in Materials and methods section, followed by colitis induction. At the end of experiments, colons from each group were collected. (a) Microscopic images of the colons are shown by hematoxylin and eosin staining. Magnification: × 100. (b) Colitis score was determined as described in Materials and methods section. Data represent the mean±s.d. of 6–8 mice per group.
DISCUSSION
TNF has a critical role in the immunopathogenesis of IBD. Little information, however, has been available so far on the precise role of TNF signaling via TNFR1 or TNFR2 in chronic intestinal inflammation. In this report, we for the first time provide evidence that TNF-dependent signal transduction via TNFR1 or TNFR2 constrains the threshold of intensity of mucosal damage during inflammation in DSS-induced chronic colitis. Lack of TNFR1 or TNFR2 dramatically exacerbated the severity of colitis. Thus, persistent targeting TNFR using antagonists (eg, Etanercept) may be detrimental for achievement of effective treatment on patients with IBD, especially UC.
Our conclusions are in agreement with a previous study that DSS-induced inflammation was significantly enhanced in TNF-α−/− mice.29 Of note, our previous study has showed that in the course of acute intestinal inflammation, TNF signaling via TNFR1 or TNFR2 had contrasting effects.13 TNFR1 deficiency exacerbated the signs of colitis while TNFR2 ablation attenuated mucosal injury. Thus, it is plausible that TNF signaling via TNFR2 have distinct effects on the acute phase and long-term phase of intestinal inflammation, which may be attributed to the different cell types in charge of delivering TNFR2-mediated signals at different stages of inflammation. In fact, although TNFR2 is broadly expressed by colonic epithelial cells and lamina propria (LP) immune cells, TNFR2-dependent signaling in LP immune cells had dominant role during acute inflammation,13 whereas its pro-apoptotic signaling in epithelial cells might be crucial in the progression of chronic colitis as shown in this paper. On the other hand, the role of TNFR1-mediated signaling in intestinal inflammation also remains to be confusing. Popivanova et al reported that blockade of TNFR1 signaling in infiltrating myeloid cells reduced the severity of colitis and the development of colitis-associated carcinoma.30 This viewpoint is recently challenged by a study from Fortgang’s lab that signaling via TNFR1 is protective from injury and inflammation in experimental colitis,31 which is consistent with our conclusions. Thus, it is reasonable to carefully interpret the findings of TNFR roles in colitis when disease models and rodent strains used are distinguished.
In regard to underlying mechanisms, we found that the harmful outcome of lacking TNFR1 or TNFR2 on DSS-induced chronic colitis is tightly related to elevated production of proinflammatory cytokines in the inflamed tissues. Thus, during chronic intestinal inflammation, interference with TNF signaling enhances other putatively colitis-related cytokine expression, for example IL-6. This result may be somewhat surprising, given that TNF acts as an inducer for IL-6 production through NF-κB-, and AP-1-dependent mechanisms,32 and it also can favor the recruitment and survival of proinflammatory immune cells capable of producing IL-6.3 Of note, in the animal model acute colitis, TNFR1 deficiency led to significant upregulation of IL-6 expression in the colon, while TNFR2 knockout did not. Therefore, elevated levels of several proinflammatory cytokines in the lesions may give an explanation for chronic colitis phenotype in TNFR1 or TNFR2−/− mice, although the underlying mechanisms remain elusive.
One of the hallmarks of intestinal inflammation is the infiltration of neutrophils into colons and release of large amounts of oxygen radicals.18 Our results showed significant increase of neutrophil infiltrates in sites of inflammation in TNFR1−/− and TNFR2−/− mice after DSS, because of upregulated expression of several well-recognized neutrophil-chemoattractant chemokines in local compartment.33 Thus, we believe that signaling via TNFR1 or TNFR2 has potentials to limit the recruitment of neutrophils into inflammatory sites during chronic intestinal inflammation and lack of TNF signaling via TNFR1 or TNFR2 facilitates the recruitment of neutrophils into inflamed tissues and ensuing boosted release of proinflammatory mediators, finally leads to aggravated signs of colitis.
Disruption of epithelial cells is a key event for DSS-induced colonic inflammation. Our results showed that the establishment of chronic intestinal inflammation led to apoptosis of colonic epithelial cells. Lack of TNF signaling via TNFR1 or TNFR2 could accelerate this apoptosis-promoting process and rendered increased death of epithelial cells by enhancing pro-apoptotic signaling and upregulating caspase-8 expression. Considering that several proinflammatory cytokines, which are well known to have potentials to promote apoptosis of epithelial cells,34, 35, 36 are highly expressed in the attacked tissues, it may be accepted that signaling via TNFR1 or TNFR2 deficiency rendered upregulation of the expression of proinflammatory mediators in the colons, the latter provoke apoptosis-promoting pathways in epithelial cells. Although the factors and their pathways involved in enhanced apoptosis of TNFR1 or TNFR2−/− null epithelial cells during chronic colitis need to be further defined, the present data indicate that increased apoptosis of colonic epithelial cells is one of the reasons for exacerbated inflammation in TNFR1 or TNFR2−/− mice after DSS.
Intriguingly, although the magnitude of local inflammatory response is comparable between TNFR1 and TNFR2−/− mice after DSS, TNFR1 ablation resulted in augmented systemic inflammatory response by upregulating GMCSF expression, which might account for more mortality in TNFR1−/− colitis mice. It has been reported that in the settings of systemic inflammatory response syndrome, circulating GMCSF has the ability to delay neutrophil apoptosis by inhibition of the generation of reactive oxygen species.37 Thus, the actions of GMCSF during chronic inflammation in TNFR1−/− mice likely include promoting myelopoiesis and prolonging survival of inflammatory cells. Moreover, our current study suggests that during chronic inflammation, TNF induces GMCSF expression via TNFR2, which is counteracted by signaling via TNFR1. Notably, GMCSF appears to be an important factor for epithelial survival, as GMCSF−/− mice developed more severe colitis in response to enteric exposure to DSS,38 which is inconsistent with the data presented in this study that blocking GMCSF failed to affect the severity of mucosal damage. This discrepancy may be resolved by the fact that at the mucosal sites of overt inflammation in TNFR1−/− colitis mice, biological function of GMCSF may be redundant. Overall, these studies indicate diverse effects of GMCSF in local and systemic inflammation.
Taken together, in the current study, we delineated a protective role of TNFR1 or TNFR2 in the course of chronic DSS-induced colitis, which is associated with alteration in several parameters, including infiltrates of granulocytes, release of proinflammatory mediators and apoptosis of epithelial cells. These data suggest that persistent and robust targeting TNF or its receptor might be have detrimental influence on the course of disease in patients with UC. Indeed, clinical observations have evidenced increased risk of IBD in patients with juvenile idiopathic arthritis treated with etanercept.39 Our current data may partially account for this confusion. Furthermore, administration of antagonists targeting TNFR1 specifically may lead to increased mortality in recipients.40 Thus, in patients with chronic inflammatory diseases, the treatment with anti-TNF or related antagonists must be strictly manipulated to avoid life-threatening side effects.
References
Hehlgans T, Pfeffer K . The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology 2005;182:1281–1290.
Papadakis KA, Targan SR . Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med 2000;51:289–298.
Kallias G, Douni E, Kassiotis G et al. On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immuol Rev 1999;169:175–194.
MacEwan DJ . TNF receptor subtype signaling: differences and cellular consequences. Cell Signal 2002;14:477–492.
Pimental-Munios FX, Seed B . Regulated commitment of TNF signaling: a molecular switch for death or activation. Immunity 1999;11:783–793.
Holtmann MH, Douni E, Schutz M et al. Tumor necrosis factor receptor 2 is up-regulated on lamina propria T cells in Crohn’s disease and promotes experimental colitis in vivo. Eur J Immunol 2002;32:142–151.
Mizoguchi E, Mizoguchi A, Takedatsu H et al. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology 2002;122:134–144.
Sashio H, Tamura K, Ito R et al. Polymorphisms of the TNF gene and the TNF receptor superfamily member 1B gene are associated with susceptibility to ulcerative colitis and Crohn’s disease, respectively. Immunogenetics 2002;53:1020–1027.
Schneider JD, Seibold I, Saxer-Sekulic N et al. Lack of TNFR2 expression by CD4+T cells exacerbates experimental colitis. Eur J Immunol 2009;39:1743–1753.
Ebach DR, Newberry R, Stenson W . Differential role of tumor necrosis factor receptors in TNBS colitis. Inflamm Bowel Dis 2005;11:533–540.
Nakai M, Sudo K, Yamada Y . The role of the tumor necrosis factor receptor in 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis in mice. Dig Dis Sci 2005;50:1669–1676.
Yang Y, Wang H, Dou Y et al. Colitogenic role of tumour necrosis factor (TNF) receptors in trinitrobenzene sulphonic acid colitis: TNF-R1 ablation does not affect systemic inflammatory response. Clin Exp Immunol 2011;165:372–382.
Wang K, Han G, Dou Y et al. Opposite role of tumor necrosis factor receptors in dextran sulfate sodium-induced colitis in mice. PLoS One 2012;7:e52924.
Zhao X, Mohaupt M, Jiang J et al. Tumor necrosis factor receptor 2-mediated tumor suppression is nitric oxide dependent and involves angiostasis. Cancer Res 2007;67:4443–4450.
Scheiffele F, Fuss I . Induction of TNBS colitis in mice. Curr Protoc Immunol 2002;S45:15.19.1–15.19.14.
Guo J, Longshore S, Nair R et al. Retinoblastoma protein (pRb), but not p107 or p130, is required for maintenance of enterocyte quiescence and differentiation in small intestine. J Biol Chem 2009;284:134–140.
Rachmilewitz D, Simon PL, Schwartz LW et al. Inflammatory mediators of experimental colitis in rats. Gastroenterology 1989;97:326–337.
Krawisz JE, Sharon P, Stenson WF . Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology 1984;87:1344–1350.
Mantovani A, Cassatella MA, Costantini C et al. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011;11:519–531.
Grisham MB, Yamada T . Neutrophils, nitrogen oxides, and inflammatory bowel disease. Ann N Y Acad Sci 1992;664:103–115.
Lochman I, Kral V, Lochmanova A et al. ANCA in the diagnosis of neutrophil-mediated inflammation. Autoimmun Rev 2011;10:295–298.
Yamaoka T, Yan F, Cao H et al. Transactivation or EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc Natl Acad Sci USA 2008;105:11772–11777.
Fish RJ, Kruithof EK . Evidence for serpinB2-independent protection from TNF-α-induced apoptosis. Exp Cell Res 2006;312:350–361.
Crespo I, San-Miguel B, Prause C et al. Glutamine treatment attenuates endoplasmic reticulum stress and apoptosis in TNBS-induced colitis. PLoS One 2012;7:e50407.
Satoh Y, Ishiguro Y, Sakuraba H et al. Cyclosporine regulates intestinal epithelial apoptosis via TGF-beta-related signaling. Am J Physiol Gastrointest Liver Physiol 2009;297:G514–G519.
Jones SA, Butler RN, Sanderson IR et al. The effect of specific caspase inhibitors on TNF-alpha and butyrate-induced apoptosis of intestinal epithelial cells. Exp Cell Res 2004;292:29–39.
Sydora BC, Albert EJ, Foshaug RR et al. Intravenous injection of endogenous microbial components abrogates DSS-induced colitis. Dig Dis Sci 2012;57:345–354.
Westbrook AM, Schiestl RH . Atm-deficient mice exhibit increased sensitivity to dextran sulfate sodium-induced colitis characterized by elevated DNA damage and persistent immune activation. Cancer Res 2010;70:1875–1884.
Barreda DR, Hanington PC, Belosevic M . Regulation of myeloid development and function by colony stimulating factors. Dev Comp Immunol 2004;28:509–554.
Naito Y, Takagi T, Handa O et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J Gastroenterol Hepatol 2003;18:560–569.
Popivanova BK, Kitamura K, Wu Y et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest 2008;118:560–570.
Chang F, Lacey MR, Bouljihad M et al. Tumor necrosis factor receptor 1 functions as a tumor suppressor. Am J Physiol Gastrointest Liver Physiol 2012;302:G195–G206.
Legrand-Poels S, Schoonbroodt S, Piette J . Regulation of interleukin-6 gene expression by pro-inflammatory cytokines in a colon cancer cell line. Biochem J 2000;349:765–773.
Kobayashi Y . The role of chemokines in neutrophil biology. Front Biosci 2008;13:2400–2407.
Tu SP, Quante M, Bhagat G et al. IFN-γ inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res 2011;71:4247–4259.
Usuda J, Okunaka T, Furukawa K et al. Increased cytotoxic effects of photodynamic therapy in IL-6 gene transfected cells via enhanced apoptosis. Int J Cancer 2001;93:475–480.
Fanning NF, Kell MR, Shorten GD et al. Circulating granulocyte macrophage colony-stimulating factor in plasma of patients with the systemic inflammatory response syndrome delays neutrophil apoptosis through inhibition of spontaneous reactive oxygen species generation. Shock 1999;11:167–174.
Xu Y, Hunt NH, Bao S . The role of granulocyte macrophage-colony-stimulating factor in acute intestinal inflammation. Cell Res 2008;18:1220–1229.
van Dijken TD, Vastert SJ, Gerloni VM et al. Development of inflammatory bowel disease in patients with juvenile idiopathic arthritis treated with etanercept. J Rheumatol 2011;38:1441–1446.
Moore TA, Perry ML, Getsoian AG et al. Increased mortality and dysregulated cytokine production in tumor necrosis factor receptor-1-deficient mice following systemic Klebsiella pneumoniae infection. Infect Immunol 2003;71:4891–4900.
Acknowledgements
We thank Professor Zhihai Qin for supplying TNFR-knockout mice. This work was supported by grants from the National Key Basic Research Program of China (2013CB530506) and the National Natural Science Foundation of China (81272320 and 81172800).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Tumor necrosis factor (TNF) deficiency exacerbates the severity of experimental chronic colitis. TNF receptor 1 (TNFR1) −/−, but not TNFR2−/− mice, show increased mortality and augmented systemic inflammatory response compared to wild-type littermates. Granulocyte-macrophage colony-stimulating factor (GMCSF) is a key mediator in this process. Signaling via TNFR therefore plays a protective role in chronic intestinal inflammation, and the lack of TNFR1 augments a systemic inflammatory response in GMCSF-dependent manner.
Rights and permissions
About this article
Cite this article
Wang, Y., Han, G., Chen, Y. et al. Protective role of tumor necrosis factor (TNF) receptors in chronic intestinal inflammation: TNFR1 ablation boosts systemic inflammatory response. Lab Invest 93, 1024–1035 (2013). https://doi.org/10.1038/labinvest.2013.89
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2013.89
Keywords
This article is cited by
-
Inflammation-induced JMJD2D promotes colitis recovery and colon tumorigenesis by activating Hedgehog signaling
Oncogene (2020)
-
Novel TNF receptor-1 inhibitors identified as potential therapeutic candidates for traumatic brain injury
Journal of Neuroinflammation (2018)
-
NLRP6 function in inflammatory monocytes reduces susceptibility to chemically induced intestinal injury
Mucosal Immunology (2017)
-
Sexually dimorphic characteristics of the small intestine and colon of prepubescent C57BL/6 mice
Biology of Sex Differences (2014)
