
Abstract
This paper describes the development of a microfluidic methodology, using RNA extraction and reverse transcription PCR, for investigating expression levels of cytochrome P450 genes. Cytochrome P450 enzymes are involved in the metabolism of xenobiotics, including many commonly prescribed drugs, therefore information on their expression is useful in both pharmaceutical and clinical settings. RNA extraction, from rat liver tissue or primary rat hepatocytes, was performed using a silica-based solid-phase extraction technique. Following elution of the purified RNA, amplification of target sequences for the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and the cytochrome P450 gene CYP1A2, was carried out using a one-step reverse transcription PCR. Once the microfluidic methodology had been optimized, analysis of control and 3-methylcholanthrene-induced primary rat hepatocytes were used to evaluate the system. As expected, GAPDH was consistently expressed, whereas CYP1A2 levels were found to be raised in the drug-treated samples. The proposed system offers an initial platform for development of both rapid throughput analyzers for pharmaceutical drug screening and point-of-care diagnostic tests to aid provision of drug regimens, which can be tailor-made to the individual patient.
Similar content being viewed by others
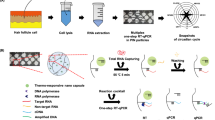
Main
Cytochrome P450 enzymes (P450s) are a superfamily of haem-containing monooxygenase enzymes, which have specific roles in numerous metabolic and synthetic pathways.1 P450s are the major enzymes involved in phase I metabolism of xenobiotics, ie, any foreign chemicals that can be introduced into an organism, including deliberately administered pharmaceutical agents.2 Phase I metabolism involves the oxidation, hydroxylation, reduction, and hydrolysis of drug molecules making them more water soluble for easy removal from the body. P450 enzymes, as well as metabolizing drugs, are also involved in drug–drug interactions, in which induction or inhibition of P450 expression can result in a greater production of toxic metabolites, or a decrease in efficacy of a drug due to increased clearance.1 There are situations in which, instead of assisting in the elimination of drugs, P450s can activate prodrugs or procarcinogens, causing them to become toxic.3 This is the case with polycyclic aromatic hydrocarbons (PAHs), which are found in compounds such as tar. PAHs by themselves cannot damage DNA, but once hydroxylated by P450 they become activated and have a carcinogenic role.4
Measuring P450 gene expression levels can be beneficial in the development and administration of pharmaceutical drugs. For example, the levels of P450 enzymes found in cells after exposure to a new drug could indicate the likely outcome of that drug due to a fast metabolism of the drug, or its conversion to undesirable by-products. Specifically, an individual patient’s likely response to a particular drug could be determined before use through testing on a biopsy taken from the patient. A tailored dosage could then be administered taking into consideration the metabolic effects of the P450 enzymes, and reducing the risk of any adverse effects to the patient.5
CYP1A2 is a cytochrome P450 enzyme associated with the metabolism of a number of clinically relevant drugs, such as clozapine (an antipsychotic), which is released from the cell under the induction of various chemicals. One way in which P450 induction can be assessed is via the 7-ethoxyresorufin O-dealkylation (EROD) assay.6 In this assay, 7-ethoxyresorufin is converted, by CYP1A2, to resorufin that can then be quantified fluorescently.7 Hepatotoxicity studies have been carried out previously within a microfluidic environment using in situ quantification of the fluorescent intensity of cells and their surrounding media via the EROD assay to compare CYP1A2 expression in control and 3-methylcholanthrene (3-MC)-induced primary rat hepatocytes.8 Assessment of the catalytic action of CYP1A2 provides information as to the action of the protein but does not measure gene expression levels. Information about changes to specific gene expression levels, in response to certain conditions, can only be obtained by analyzing levels of messenger RNA (mRNA).
To obtain this information, mRNA must first be extracted from the biological matrix that is being investigated. The purified mRNA is then converted to complementary DNA (cDNA) by a reverse transcriptase enzymatic reaction. The cDNA can then be amplified using the PCR, which enables either end-point or real-time detection.
The increasing ability to miniaturize laboratory techniques onto a ‘Lab-on-a-Chip’ format using microfluidic technology has opened the door to many varied and far-reaching applications. It is now possible to integrate reactions in a way not possible before, so that instead of products being moved from one reaction vessel to another, risking contamination and loss of product, two or more reactions can happen on the same enclosed microfluidic device.9
Although nucleic acid analysis on microfluidic devices has received great interest in the literature, the majority of publications are focussed on DNA presumably because of the less-stable nature of RNA and its susceptibility to RNases. However, analysis of RNA is vital for gene expression studies. Total RNA can be extracted from biological samples using a solid-phase matrix to which nucleic acids bind under selective conditions. The most common methods involve either a silica- or anion exchange-based solid phase. When using silica, nucleic acids bind to the solid phase in the presence of a chaotropic salt such as guanidine hydrochloride, and unbound contaminants are then removed with an alcohol wash before the purified nucleic acids are eluted in a low ionic strength buffer.10 Chitosan is an example of an anion exchange matrix that can be used to bind nucleic acids in solution at pH 5 and elute them at pH 9 once unbound contaminants have been removed.11 Alternatively, mRNA can be isolated via the poly(A) tail using tethered poly(T) sequences, eg, directly immobilized onto the channel surface12 or on superparamagnetic beads enabling the captured mRNA sequences to be manipulated around the microfluidic device using an external magnet.13 More recent developments have enabled even more selective extraction of RNA using sequence-specific probes. Root et al14 presented an innovative method for the purification of RNA from serum using a polymer capture matrix containing covalently bound oligonucleotides. The capture, wash, and elution of target RNA sequences were controlled by electrophoresis, with an additional heating step required for elution.
A variety of amplification methods have been applied to microfluidic devices to analyze RNA. Amplification of RNA viruses, as well as the more traditional single chamber for aqueous RT-PCR, has been achieved using both continuous flow15 and solid-phase microarrays.16 Isothermal techniques, such as nucleic acid sequence-based amplification17 and loop-mediated isothermal amplification,18 have also been successfully used.
The integration of RNA purification and amplification processes is a challenge that has led to the development of a number of inventive solutions. For example, Pipper et al19 used a series of aqueous droplets in oil on a perfluorinated surface. Silica-coated superparamagnetic particles were incubated with a biological sample and then dragged through droplets containing wash and elution reagents for RT-PCR before amplification and real-time detection. Lateral flow strips have also been incorporated into a microfluidic device for detection of RT-PCR products for HIV.20 Amplicons are labeled with digoxigenin and biotin that bind to immobilized streptavidin and are then detected using up-converting phosphor reporter particles. Another alternative approach was presented by Ferguson et al21 who used immunomagnetic target capture combined with sequence-specific electrochemical detection for identification of the H1N1 influenza virus from throat swab samples within 3½ h.
Microfluidic devices provide a biomimetic microenvironment that allows tissue samples/biopsies to be maintained for up to 8 days.22, 23 This enables investigation of the tissue as a whole or of individual cells following disaggregation.24 Here we demonstrate the integration of RNA extraction with RT-PCR on a single microfluidic device that could be used for the analysis of tissue samples. CYP1A2 gene expression levels, from 3-MC-induced and non-induced cell populations, were compared with gene expression levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control.
MATERIALS AND METHODS
Preparation of Microfluidic Devices
The microfluidic device used was made of borosilicate glass and prepared using standard photolithography and wet etching techniques to produce the design shown in Figure 1. The etched bottom plate was then thermally bonded to a top plate containing 360 μm holes drilled to act as inlets and outlets for the channels.

Schematic diagram of the microfluidic device used for the integration of RNA extraction and RT-PCR. All features were etched to a depth of 500 μm. (a) Monolith solution inlet. (b) Sample, wash and elution reagent inlet. (c) Waste outlet. (d) PCR reagent inlet. (e) Waste outlet.
The internal surfaces of the microfluidic device were silanized to minimize DNA polymerase adsorption. The channels were washed overnight with 10% (w/v) NaOH, then sequentially with 10 ml water and ethanol before being dried thoroughly in an oven at 90 °C. Sigmacote (Sigma-Aldrich, UK) was then applied to the channels for 5 min, following which the microfluidic devices were dried in an oven for 30 min. Porous thermally activated silica monoliths were generated in the RNA-extraction chamber. The monoliths were made from potassium silicate (K2SiO3 (21% SiO2, 9% K2O)) (VWR International, UK) and formamide (Alfa Aesar, UK) in a 10:1 ratio, and cured in an oven overnight at 90 °C.25 The monoliths generated are contained within the hexagonal RNA-extraction chamber. Porous silica monoliths have been shown to contain both micron and nm-scale pores, resulting in facile flow of solutions through the micron pores and generation of a large surface area for reactions due to the nm-scale pores.26 In this instance, the micron pores have an average diameter of 1.6 μm (Supplementary Material, Supplementary Figure S1). Agarose gel encapsulated RT-PCR reagents were then filled into half of the PCR chamber and the microfluidic device stored at 4 °C until required.27
Sample Preparation
Experiments used either rat liver tissue or primary rat hepatocytes. Male Wistar rats were housed under standard conditions. All animals were fed and watered ad libitum until anaesthetized and killed under schedule 1 procedure for liver extraction. Total hepatectomy was performed, and tissue sections of approximately 1 mm3 were stabilized in RNAlater solution (Qiagen, UK) to prevent degradation of RNA and were stored at −20 °C. Control and 3-MC-induced primary rat hepatocytes were provided by LGC (Teddington, UK) and were stored in Trizol (Invitrogen, UK) at −80 °C until required.
Conventional RNA Extraction
All solutions were prepared using diethylpyrocarbonate (Sigma-Aldrich) treated water. RNA extraction was carried out from the stabilized liver tissue, following the Qiagen RNeasy Mini Kit (Qiagen) protocol. Briefly, tissue sections were homogenized in RLT buffer supplemented with 2 M dithiothreitol (Fluka, UK) using a 1 ml syringe and a needle. Homogenized cells were applied to an RNeasy spin column, washed, and then eluted in 50 μl of water and stored at −20 °C until required. RNA extraction from primary rat hepatocytes (∼0.4 × 106 cells) was carried out using conventional Trizol methodology.
Microfluidic RNA Extraction
Before sample addition, the monoliths were pretreated with 10 mM TE buffer (10 mM Tris, 1 mM EDTA, adjusted to pH 6.7), which was hydrodynamically pumped through at 5 μl/min. Rat liver tissue (1 mm3) or primary hepatocytes (∼0.4 × 106 cells) were homogenized in 40 μl 5 M guanidine hydrochloride (Sigma-Aldrich) in 10 mM TE buffer, and then pumped through the monolith at 2.5 μl/min. The optional addition of DNase I (Invitrogen) to this lysis/binding buffer was also evaluated. A 50 μl solution of 80% (v/v) isopropanol (Sigma-Aldrich) was then used to wash the monolith and remove cellular debris and potential contaminants of downsteam processes. The concentrated RNA was eluted from the monolith using 50 μl of water at a flow rate of 1 μl/min. Fractions of 5 μl were continuously collected throughout the extraction procedure for RNA quantification and downstream analysis using RT-PCR.
RNA Quantification
RNA quantification was carried out using a Nanodrop spectrometer (ThermoScientific, UK) using 1 μl aliquots taken from each collected fraction.
RT-PCR
Intron spanning primers were used for GAPDH and CYP1A2 to facilitate amplification of transcribed mRNA and not genomic DNA (Table 1). GAPDH primers were previously published,1 whereas CYP1A2 primers were designed using Primer-BLAST software and synthesized de novo. GAPDH, a housekeeping gene, was used as a positive control because of its constitutive expression.
A one-step RT reaction and PCR were combined for generation of cDNA and subsequent amplification. RT-PCR was carried out using the following reagent mixture: 2.5 μM forward primers, 2.5 μM reverse primers, 0.5 μM probes, 1 × M-MLV reaction buffer, 200 U M-MLV reverse transcriptase, 200 μM each deoxyribonucleotides, 2 mM MgCl2, 20 μg/ml bovine serum albumin, and 0.2 U GoTaq DNA polymerase (Promega, UK).
After the RNA was eluted from the monolith, the inlets/outlets of the microfluidic device were covered with a drop of mineral oil to prevent evaporation of reagents during thermal cycling. The microfluidic device was placed on a thermoelectric Peltier element, which provided heating and cooling for RT-PCR. Control samples were run on a conventional TC-312 (Techne, UK) thermal cycler. All samples were run under the following conditions: reverse transcription (70 °C for 5 min, 4 °C for 30 s, and 37 °C for 15 min) and PCR (28 cycles of 94 °C for 30 s, 59 °C for 30 s, and 72 °C for 30 s). Amplified samples were analyzed either by agarose gel electrophoresis or capillary gel electrophoresis.
Gel Electrophoresis
Products of RT-PCR were analyzed using agarose gel electrophoresis. Agarose gels of 2% (w/v) concentration were made in 0.5 × Tris/Borate/EDTA buffer. Loading dye was added to the RT-PCR products before being run at 120 V for 90 min alongside Hyperladder II (Bioline, UK) for comparison. Ethidium bromide at a concentration of 0.5 μg/ml (CLP, US) was used to stain the DNA for visualization using a UV transilluminator.
RT-PCR products were alternatively analyzed using a standard procedure on a Capillary Gel Electrophoresis CEQ 8000 Genetic Analyser (Beckman-Coulter, UK).
RESULTS
RNA Extraction
As silica is known to bind all nucleic acids, DNase I was used to remove any potentially contaminating DNA and maximize RNA-extraction efficiency. This was carried out by adding DNase I, at a range of 0.01–10 mg/ml, to the binding buffer. Optimization of this process showed that a concentration of 1 mg/ml removed the maximum amount of DNA (data not shown).
Preconditioning of the silica monolith using 10 mM TE buffer, pH 6.7, was performed in order to confer the most appropriate level of protonation to the surface in order to maximize nucleic acid adsorption/desorption28 A 30-min conditioning step was found to be optimal as it resulted in maximal RNA yields during the elution phase, approximately 32% greater total yield than with no preconditioning (Figure 2). Longer incubations times, 60 min, resulted in loss of RNA during the washing phase, suggesting an inefficient binding process.

Graph showing an example of the RNA-elution profile results obtained when the monolith was subjected to different preconditioning times of 0 (—▪—), 30 (---○---), and 60 min (···Δ···) (n=3 for each condition tested).
Following optimization of the RNA-extraction process on the proposed microfluidic system, whole tissue was homogenized directly in the lysis/binding buffer and RNA extracted. RNA was successfully extracted from rat liver tissue using this technique, and the eluted fractions are shown to be of sufficient quantity and quality for amplification using RT-PCR on the microfluidic device (Figure 3).

Graph showing average elution profile from RNA extracted from rat liver tissue on a microfluidic device. *Denotes samples that were successfully amplified by RT-PCR (n=3).
RT-PCR
Combined RT-PCR for multiplex amplification was optimized on the benchtop and then transferred onto the microfluidic system. Successful amplification of both targets was demonstrated on the microfluidic device as demonstrated by the generation of PCR products of the expected sizes for GAPDH and CYP1A2 (Figure 4).

Gel electropherogram showing RT-PCR products: (1) DNA size ladder; (2) positive control samples amplified on a conventional thermal cycler; (3) RT-PCR samples amplified on the microfluidic device, GAPDH (91 bp), and CYP1A2 (129 bp); and (4) negative control (n=3).
Process Integration on a Microfluidic Device
To facilitate integration of the RNA extraction and RT-PCR process on a single microfluidic device, the RT-PCR reagents were encapsulated in a 1.5% (w/v) agarose gel and filled into half of the amplification chamber. Once the RNA was eluted from the monolith, the flow was directed toward the amplification chamber, whereupon it filled the second half of the amplification chamber. The inlets and outlets of the microfluidic device were then covered with a layer of mineral oil to prevent evaporation. Upon the initial reverse transcription heating step, the agarose gel melts to release the RT-PCR reagents, which combine with the eluted RNA. Following amplification on the microfluidic device, RT-PCR products were analyzed by capillary gel electrophoresis, and the relative fluorescent intensities of the products were compared to enable qualitative analysis of the end-point RT-PCR. This technique was successfully used to compare basal gene expression levels of CYP1A2 with those induced by 3-MC, using GAPDH as a constitutively expressed control (Figure 5). As expected, treatment with 3-MC led to induction of CYP1A2 resulting in increased gene expression levels; P=0.008, two-tailed paired t-test, which was in agreement with Baldwin et al.1

Comparison of 3-MC-induced and non-induced primary rat hepatocytes as extracted and amplified for GAPDH (black) and CYP1A2 (gray) using the integrated microfluidic system (n=3). A statistically significant difference in CYP1A2 induction by 3-MC was observed.
DISCUSSION
The ability to monitor drug effects on tissue samples using a microfluidic system offers numerous advantages over conventional drug-examination methods. First, multiple experiments could be carried out on a single biopsy sample, reducing the number of animals required for basic research or enabling a patient biopsy to be used enabling individually tailored medications and treatment plans to be devised.5, 29 Furthermore, as the effect of the drug can be monitored not only at an observable cellular level but also at the level of gene expression, the pharmacological effect can be monitored looking at potential unwanted side effects caused by the activation of prodrugs or carcinogens. Integration of the proposed system with other techniques, such as the EROD assay, would allow more information to be obtained from individual biopsy samples, eg, protein activity and gene expression levels, in the development of techniques for patient-specific treatments. Most important is the ability to integrate analysis directly from a biopsy of tissue. This allows direct interrogation in the most appropriate manner depending on the type of analysis to be performed, allowing extraction and quantification of the mRNA within approximately 100 min from the time of sample input into the microfluidic device. Integration minimizes contamination and loss of sample, making the whole process more reliable and applicable to small samples—useful in many clinical settings where the tissue is often limited. In addition to measuring changes in mRNA expression, the tissue-based microfluidic system can be coupled to other ‘downstream’ analysis modules, eg, spectrophotometric/fluorescent detection of protein release,30 individual cells can be liberated and changed to the cell surface analyzed by on-chip flow cytometry,24 and oligonucleotide hybridization arrays can be used to assess global changes in gene expression.31 The combination of these technologies together with the RT-PCR described here has the potential to offer a new platform technology for studying normal or diseased tissue.
References
Baldwin SJ, Bramhall JL, Ashby CA et al. Cytochrome P450 gene induction in rats ex vivo assessed by quantitative real-time reverse transcriptase-polymerase chain reaction (taqman). Drug Metab Dispos 2006;34:1063–1069.
Ek M, Soderdahl T, Kuppers-Munther B et al. Expression of drug metabolizing enzymes in hepatocyte-like cells derived from human embryonic stem cells. Biochem Pharmacol 2007;74:496–503.
Baudoin R, Corlu A, Griscom L et al. Trends in the development of microfluidic cell biochips for in vitro hepatotoxicity. Toxicol In Vitro 2007;21:535–544.
Xue W, Warshawsky D . Metabolic activation of polycyclic and heterocyclic aromatic hydrocarbons and DNA damage: A review. Toxicol Appl Pharmacol 2005;206:73–93.
Bates S . Progress towards personalized medicine. Drug Discov Today 2010;15:115–120.
Vakharia DD, Liu N, Pause R et al. Effect of metals on polycyclic aromatic hydrocarbon induction of CYP1A1 and CYP1A2 in human hepatocyte cultures. Toxicol Appl Pharmacol 2001;170:93–103.
Kennedy SW, Jones SP, Bastien LJ . Efficient analysis of cytochrome P4501A catalytic activity, porphyrins, and total proteins in chicken embryo hepatocyte cultures with a fluorescence plate reader. Anal Biochem 1995;226:362–370.
Anderson K, Cooper JM, Haswell SJ et al. Microfluidic-based measurements of cytochrome P450 enzyme activity of primary mammalian hepatocytes. Analyst 2010;135:1282–1287.
Whitesides GM . The origins and the future of microfluidics. Nature 2006;442:368–373.
Wang C, Kim T, Gao D et al. Rapid high-yield mRNA extraction for reverse-transcription PCR. J Biochem Biophys Methods 2006;70:507–509.
Hagan KA, Meier WL, Ferrance JP et al. Chitosan-coated silica as a solid phase for RNA purification in a microfluidic device. Anal Chem 2009;81:5249–5256.
Hughes-Chinkhota CN, Banda M, Smolinski JM et al. Oligonucleotide immobilization using 10-(carbomethoxy)decyl- dimethylchlorosilane for mRNA isolation and cDNA synthesis on a microfluidic chip. Sensors Actuators B: Chem 2011;155:437–445.
Jiang GF, Harrison DJ . mRNA isolation in a microfluidic device for eventual integration of cDNA library construction. Analyst 2000;125:2176–2179.
Root BE, Agarwal AK, Kelso DM et al. Purification of HIV RNA from serum using a polymer capture matrix in a microfluidic device. Anal Chem 2011;83:982–988.
Li Y, Zhang C, Xing D . Fast identification of foodborne pathogenic viruses using continuous-flow reverse transcription-PCR with fluorescence detection. Microfluid Nanofluid 2011;10:367–380.
Sun Y, Dhumpa R, Bang DD et al. A lab-on-a-chip device for rapid identification of avian influenza viral RNA by solid-phase PCR. Lab on a Chip 2011;11:1457–1463.
Dimov IK, Garcia-Cordero JL, O'Grady J et al. Integrated microfluidic tmRNA purification and real-time NASBA device for molecular diagnostics. Lab on a Chip 2008;8:2071–2078.
Wang CH, Lien KY, Wang TY et al. An integrated microfluidic loop-mediated-isothermal-amplification system for rapid sample pre-treatment and detection of viruses. Biosens Bioelectron 2011;26:2045–2052.
Pipper J, Inoue M, Ng LF-P et al. Catching bird flu in a droplet. Nat Med 2007;13:1259–1263.
Chen D, Mauk M, Qiu X et al. An integrated, self-contained microfluidic cassette for isolation, amplification, and detection of nucleic acids. Biomed Microdevices 2010;12:705–719.
Ferguson BS, Buchsbaum SF, Wu TT et al. Genetic analysis of H1N1 influenza virus from throat swab samples in a microfluidic system for point-of-care diagnostics. J Am Chem Soc 2011;133:9129–9135.
Hattersley SM, Dyer CE, Greenman J et al. Development of a microfluidic device for the maintenance and interrogation of viable tissue biopsies. Lab on a Chip 2008;8:1842–1846.
Hattersley SM, Sylvester DC, Dyer CE et al. A microfluidic system for testing the responses of head and neck squamous cell carcinoma tissue biopsies to treatment with chemotherapy drugs. Ann Biomed Eng 2012;40:1277–1288.
Woods J, Docker PT, Dyer CE et al. On-chip integrated labelling, transport and detection of tumour cells. Electrophoresis 2011;32:3188–3195.
Oakley JA, Shaw KJ, Docker PT et al. Development of a bi-functional silica monolith for electro-osmotic pumping and DNA clean-up/extraction using gel-supported reagents in a microfluidic device. Lab on a Chip 2009;9:1596–1600.
Fletcher PDI, Haswell SJ, He P et al. Permeability of silica monoliths containing micro- and nano-pores. J Porous Mater 2011;18:501–508.
Shaw KJ, Joyce DA, Docker PT et al. Development of a real-world direct interface for integrated DNA extraction and amplification in a microfluidic device. Lab on a Chip 2011;11:443–448.
Melzak KA, Sherwood CS, Turner RFB et al. Driving forces for DNA adsorption to silica in perchlorate solutions. J Colloid Interface Sci 1996;181:635–644.
Shaw KJ, Birch C, Hughes EM et al. Microsystems for personalized biomolecular diagnostics. Eng Life Sci 2011;11:121–132.
Martino C, Zagnoni M, Sandison ME et al. Intracellular protein determination using droplet-based immunoassays. Anal Chem 2011;83:5361–5368.
Demarest TG, Murugesan N, Shrestha B et al. Rapid expression profiling of brain microvascular endothelial cells by immuno-laser capture microdissection coupled to TaqMan ® low density array. J Neurosci Methods 2012;206:200–204.
Acknowledgements
The authors would like to thank Dr Steve Clark for fabrication of the microfluidic devices, LGC for providing rat hepatocytes, and HYMS for funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Cytochrome P450 enzymes are involved in the metabolism of xenobiotics, including many commonly prescribed drugs. This paper describes the development of a rapid throughput microfluidic methodology for investigating expression levels of cytochrome P450 genes, which may be useful for development of pharmaceutical drug screens and diagnostic tests to guide personalized drug regimens.
Supplementary information
Rights and permissions
About this article
Cite this article
Shaw, K., Hughes, E., Dyer, C. et al. Integrated RNA extraction and RT-PCR for semi-quantitative gene expression studies on a microfluidic device. Lab Invest 93, 961–966 (2013). https://doi.org/10.1038/labinvest.2013.76
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2013.76
