
Abstract
Lysophosphatidic acid (LPA) has a critical role in lymphocyte migration to secondary lymphoid organs. Autotaxin (ATX)/lysophospholipase D, in the vascular endothelium, is the main enzyme involved in LPA production. Whether ATX is involved in pathological lymphocyte migration to the inflamed mucosa has not been studied. We investigated the involvement of ATX in inflammatory bowel disease patients and two murine models of colitis. Tissue samples were obtained by intestinal biopsies from patients with Crohn’s disease and those with ulcerative colitis with informed consent. ATX immunoreactivity was colocalized with MAdCAM-1-positive high-endothelial-like vessels, close to sites of lymphocyte infiltration. Enhanced expression of ATX mRNA was observed in the inflamed mucosa from Crohn’s disease and ulcerative colitis patients. ATX mRNA expression level was remarkably higher in the actively inflamed mucosa than in the quiescent mucosa in the same patient. In the T-cell-transferred mouse model, ATX mRNA expression level gradually increased as colitis developed. In the dextran sodium sulfate mouse model, the expression level was considerably higher in colonic mucosa of chronically developed colitis than in colonic mucosa of acute colitis. Administration of an ATX inhibitor, bithionol, remarkably decreased lymphocyte migration to the intestine and ameliorated both dextran sodium sulfate-induced colitis and CD4-induced ileocolitis. In transwell assays, administration of bithionol or 1-bromo-3(s)-hydroxy-4-(palmitoyloxy) butylphosphonate (BrP-LPA) significantly decreased transmigration of splenocytes through high-endothelial-like vessels induced by TNF-α. We conclude that enhanced expression of ATX in the active mucosa has been implicated in the pathophysiology of inflammatory bowel disease through enhancing aberrant lymphocyte migration to the inflamed mucosa.
Similar content being viewed by others

Main
Lymphocyte trafficking through the intestinal mucosa has a crucial role in the pathogenesis of inflammatory bowel disease (IBD), and intestinal microcirculation is a primary target of this inflammatory response. Lymphocyte trafficking from blood to lymphoid tissue and inflammatory sites depends on the interaction between lymphocytes and specialized high endothelial venules (HEVs).1 This interaction is thought to proceed by a sequence that includes lymphocyte rolling, chemokine-mediated activation and subsequent firm adhesion, followed by lymphocyte transendothelial migration into intestinal tissues.2 The main cell surface molecules involved in this adhesion cascade are selectins, chemokines and their receptors, immunoglobulin superfamily molecules and integrins. Enhanced expression of adhesion molecules (eg, ICAM-1 and VCAM-1) on endothelial cells has been suggested to be important for mediating tissue injury during acute flares of colitis.3 However, several studies have suggested that MAdCAM-1 represents a potentially more important therapeutic target for the treatment of chronic IBD because MAdCAM-1 is upregulated mainly on mucosal endothelial cells in the gastrointestinal tract and is involved in the selective homing of α4β7-integrin, expressed primarily on memory lymphocytes, to mucosal tissue.4, 5, 6 Recently, adhesion molecule inhibitors targeting lymphocyte trafficking in the intestine have been developed. Natalizumab is a recombinant human IgG4 monoclonal antibody against α4-integrin. It inhibits both α4β7-integrin–MAdCAM-1 interactions and α4β7-integrin–VCAM-1 binding.7 We have previously reported an ameliorating effect of an antiadhesion molecule antibody in an animal model of colitis.8 However, a combination of MAdCAM-1 and L-selectin blockade attenuated intestinal injury further as compared with the effect of blockade of MAdCAM-1 alone, suggesting that blockade of two pathways, rolling and adhesion, works more efficiently.9 In addition, colitis models by adoptive transfer of CD4+CD45RBhigh cells into severe-combined immunodeficiency (SCID) mice presents colitis even when CD4+CD45RBhigh cells were isolated from β7-deficient mice.10 It suggests that β7-independent pathways also contribute to intestinal inflammation and that targeting only MAdCAM-1–β7-integrin is not sufficient to cure intestinal inflammation. Recently, Kanda et al11 reported a previously unknown mechanism that enhances lymphocyte motility and is involved in the entry of lymphocytes into secondary lymphoid organs. This mechanism is based on autotaxin (ATX), a lysophospholipase D that is secreted by HEVs. After chemokine activation, lymphocytes can bind to this enzyme through the integrin, and the enzyme product lysophosphatidic acid (LPA) is able to enhance lymphocyte motility.12, 13 Once the intestine becomes inflamed, HEV-like vessels, which appear to be phenotypically and functionally similar to HEVs, are induced in extralymphoid sites from lymphoid tissues.14 Such vessels are composed of plump endothelial cells, contain many luminal and intramural lymphocytes (presumably in the process of extravasation) and mediate in vitro lymphocyte adhesion. During chronic inflammation of the gut in IBD, areas of dense lymphocytic infiltration contain vessels with plump endothelium-expressing MAdCAM-1.14 ATX is highly expressed in HEVs in Peyer’s patch and lymph nodes.15 However, whether ATX is expressed in HEV-like vessels in the inflamed mucosa of the intestine and whether this mechanism participates in the entry of lymphocytes into inflamed mucosa remain to be elucidated. In this study, we investigated the expression of ATX in the inflamed mucosa of IBD patients. The relationship between the degree of its expression and activity of IBD was also investigated. In addition, to determine whether ATX is a possible target for IBD treatment, we investigated whether blockade of ATX ameliorates intestinal injury through inhibiting lymphocyte migration to the intestinal mucosa by using two different models of murine colitis and bEnd3 cells.
MATERIALS AND METHODS
Evaluation of ATX in Ulcerative Colitis and Crohn’s Disease Patients
Patients
In all, 215 consecutive patients previously diagnosed with IBD were included in this study. Crohn’s disease (CD) and ulcerative colitis (UC) were based on standard clinical, endoscopic, histological and radiographic criteria. The patients included 56 patients with CD (39 men and 17 women; mean age, 34.9 years; range, 20–70 years), 159 patients with UC (70 men and 89 women; mean age 45.9 years; range, 2081 years) and 20 patients with colon polyps as healthy controls (10 men and 10 women; mean age, 56.8 years; range, 4665 years). Mucosal samples from each patient were obtained from two different sites during endoscopy: from the inflamed mucosa (inflamed) and from the quiescent mucosa (quiescent). Activity of colitis in UC patients was determined by endoscopy appearance using the criteria previously reported by Matts16 (grade 1=normal; grade 2=mild granularity of the mucosa with mild contact bleeding; grade 3=marked granularity and edema of the mucosa, contact bleeding and spontaneous bleeding; grade 4=severe ulceration of the mucosa with hemorrhage). Mucosal samples from the patients with colon polyps were obtained from healthy mucosa around the polyp when endoscopic mucosal resection was carried out. The Ethics Committee of the National Defense Medical College approved the study protocol, and all controls and patients gave informed consent for inclusion in the study.
Immunohistological staining
Samples were fixed in periodate–lysine–paraformaldehyde, and immunohistochemistry was performed on cryostat sections by the labeled streptavidin biotin technique. As primary antibodies, we used mouse anti-human CD34 (clone: NU-4A1; Nichirei Bioscience, Tokyo, Japan), rat anti-human β7-integrin (clone: FIB27; BD PharMingen, San Diego, CA, USA) and rabbit anti-human ATX (clone: polyclonal IgG; Santa Cruz Biotechnology). They were observed using a confocal LASER microscope (LSM510-V3.5; Carl Zeiss).
Real-time quantitative PCR of CD and UC samples
Colonic tissues of the colon and ileum were obtained by biopsies from patients with CD, UC or colonic polyps with informed consent. Total RNA was extracted with the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. First-strand cDNA was prepared for each RNA sample using Superscript III reverse transcriptase with oligo(dT) and random primers (Invitrogen). The transcripts were quantified by real-time quantitative PCR using the ABI 7900 HF Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) and qPCR Master Mix (Eurogentec, San Diego, CA, USA) using the corresponding primer pairs for human ATX (Hs00196470:ABI) and human GUSβ (Hs99999908:ABI). mRNA levels were normalized to GUSβ.
Evaluation of ATX in Colitis Model Mice
Administration of DSS and assessment of colonic inflammation
A 8-week-old female BALB/c mice received five cycles of dextran sodium sulfate (DSS) treatment. Each cycle consisted of 3% DSS (molecular weight of 36 000–50 000; MP Biomedicals, Illkirch, France) in drinking water for 7 days, followed by a 10-day interval with normal drinking water (n=6 per group).17 Care and use of the laboratory animals were in accordance with the guidelines of National Defense Medical College’s Animal Research Committee. Under pentobarbital anesthesia, the colon was removed and fixed in 10% buffered formalin for hematoxylin and eosin (H–E) staining or in RNAlater (Applied Biosystems) for real-time quantitative PCR.
T-cell transfer model of chronic colitis
CD4+CD25− T-cell transfer-mediated colitis was performed as described previously.18 Splenocytes were harvested from wild-type BALB/c mice at 8 weeks of age. After removing red blood cells, CD4+CD25− T cells from splenocytes were isolated by magnetic bead depletion using a mouse CD4+CD25+ Treg cell isolation kit as per the manufacturer’s recommendations (Invitrogen Dynal AS, Oslo, Norway). The purified CD4+CD25− (5 × 105) cells, diluted in 200 μl of PBS, were injected intraperitoneally into female SCID recipient mice at 6 weeks of age (n=6 per group). The care and use of the laboratory animals were in accordance with the guidelines of National Defense Medical College’s Animal Research Committee. Diseased animals were killed for analysis at 6–8 weeks after transfer. Under pentobarbital anesthesia, the colon was removed and fixed in 10% buffered formalin for H–E staining or in RNAlater (Applied Biosystems) for real-time quantitative PCR.
Histology
The colons were isolated for further analysis. Macroscopic colonic damage was assessed by colon weight-to-length ratio. For histological studies, colonic tissue located precisely 4 cm above the anus was resected from the mice in all groups at necropsy. The colonic tissues were fixed in 10% phosphate-buffered formalin (Wako Chemicals, Osaka, Japan) and embedded in paraffin. Sections (4 μm) were prepared and stained with hematoxylin–eosin. The sections were scored on the basis of three parameters: severity of inflammation (0–3: none, slight, moderate and severe), depth of injury (0–3: none, mucosal, mucosal and submucosal, and transmural) and crypt damage (0–4: none, basal & frac 13; damaged, basal & frac 23; damaged, only surface epithelium intact, loss of entire crypt and epithelium). Each parameter score was multiplied by a factor reflecting percent involvement of the intestinal wall ( × 1: 0–25%; × 2: 26–50%; × 3: 51–75% and × 4: 76–100%) and summed to obtain a histopathology score (maximum of 40 possible).19 This histological analysis was performed for each colitis mouse model.
Intravital observation of lymphocyte migration to colonic microvessels
Lymphocytes were isolated from the spleen after lysing red blood cells in ammonium phosphate/chloride lysis buffer. A T-cell-rich fraction of mesenteric lymphocytes was first obtained by using a nylon wool column. The whole cell population of 1 × 108 lymphocytes, in 20 ml of RPMI medium with 1% fetal bovine serum, was incubated in 1 g of nylon wool (Kanto Kagaku, Tokyo, Japan) in a column and the flow-through fraction was designated as the T-cell-rich fraction after incubation for 1 h at 37 °C. The cell suspensions were washed and stored in RPMI 1640 medium with 5% fetal calf serum on ice until used. The lymphocytes were labeled with carboxyfluorescein diacetate succinimidyl ester as reported previously.20 The cells were resuspended in 0.2 ml of the medium and used within 30 min. Microcirculation in the colonic mucosa was observed from the mucosal surface with an inverted-type fluorescence microscope (Diaphot TMD-2 S; Nikon, Tokyo, Japan). Lymphocytes (3 × 107 dissolved in 0.3 ml) were injected into the cervical vein of each recipient mouse for 3 min. CD4+CD25− transfer mice and wild-type BALB/c mice were used as recipient mice. CD4+CD25− transfer mice were divided into two groups: a bithionol (+) group and a bithionol (−) group (n=6 per group). Bithionol (+) group mice were injected with bithionol (1.0 mg dissolved in 0.2 ml carboxymethylcellulose (CMC) solution) intraperitoneally 12 h before the experiment. Interaction of the infused lymphocytes with the colonic microvascular beds was monitored and continuously recorded for 60 min. Lymphocytes that moved outside the vessel were counted as transmigrated cells.
Inhibitory effect of ATX inhibitor in colitis model mice
To elucidate the effect of ATX on colitis development, we used bithionol (Sigma-Aldrich) as an ATX inhibitor.21 We examined the effects of ATX inhibition in both DSS colitis model mice and CD4+CD25− T-cell transfer colitis model mice. In separate sets of experiments, treatment group mice were injected with bithionol (1 mg per body) dissolved in 0.2 ml CMC solution (0.25%) intraperitoneally every other day from the beginning of colitis induction, and vehicle control group mice were injected with 0.2 ml CMC solution (0.25%) intraperitoneally every other day from the beginning of colitis induction (n=6 per group). The effect of ATX inhibition on colitis development was assessed histologically in both models of colitis. The number of infiltrating lymphocytes per millimeter muscularis mucosa was also compared with healthy mice without bithionol group (n=6) and healthy mice without bithionol group (n=6). CD3+ cells were counted as lymphocytes.
Real-time quantitative PCR of murine samples
Total RNA was extracted from colonic samples using the RNeasy Mini isolation kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol as described above. For quantitative RT-PCR of murine intestinal mucosa, the same procedure as that using Mm0051657 for murine ATX was used. To standardize the quantitation, the TaqMan GAPDH Control Reagents kit was used.
Effect of ATX Blockade on Lymphocyte Migration Through HEVs In Vitro
As bioactive LPA itself is difficult to measure, we investigated the blocking effects of an LPA analog and an ATX inhibitor on lymphocyte transmigration through induced HEVs in vitro. Transmigration was studied, as described previously with minor modifications,22 using a modified Boyden chamber assay with 6-Well Millicell Hanging Cell Culture Inserts (Millipore Corporation, Billerica, MA, USA) containing a permeable support insert with a pore size of 5 μm. Briefly, 5 × 104 mouse bEnd3 cells (passages 6–10; ATCC) were seeded onto a Transwell insert and cultured to confluency for 2 days at 37 °C and 5% CO2. The cell monolayers were treated with 5 ng of TNF-α (Roche Diagnostics, Indianapolis, IN, USA) per well for an additional 4 h. Quantitative RT-PCR showed upregulated ATX mRNA levels in the cell monolayers (Figure 8a). Splenocytes (1 × 106) were added to the upper chamber and allowed to transmigrate for 4 h to the lower chamber. We used bithionol as an ATX inhibitor and 1-bromo-3(s)-hydroxy-4-(palmitoyloxy) butylphosphonate (BrP-LPA) as a competitive inhibitor of LPA.23, 24 In the ATX inhibition group, 10 nM bithionol per well was added to the medium. For an LPA analog, BrP-LPA (Echelon Biosciences, Salt Lake City, UT, USA) was added at a concentration of 40 μM. Cells that had transmigrated were counted using a Burker–Turk hemocytometer and analyzed by flow cytometry.
Flow cytometry
In the transwell assay mentioned above, flow cytometry analysis was also performed for cells that had transmigrated. As primary monoclonal antibodies, we used rat anti-mouse CD11b (clone: Mac-1α; BD PharMingen) and rat anti-mouse β7-integrin (clone: FIB21; BD PharMingen). The cells were analyzed using a FACS calibur flow cytometer (BD Biosciences). Data were analyzed using the CellQuest software (BD Biosciences).
Statistical Analysis
All results are expressed as means±s.e.m. Differences between two groups were evaluated using the Mann–Whitney U-test. Statistical significance was set at P<0.05. Differences between more than two groups were evaluated using one-way ANOVA and a post hoc test. P<0.05 was considered significant.
RESULTS
Evaluation of ATX in UC and CD Patients
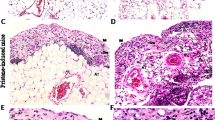
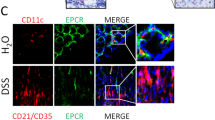
To examine the expression of ATX transcripts in inflamed colonic mucosa of IBD patients, we carried out immunohistochemical analysis to determine ATX protein localization in these tissues. To assist the identification of HEV-like vessels in inflamed intestinal mucosa, we used CD34.14 In the inflamed colonic mucosa, positive immunoreactivity of CD34 was observed in HEV-like vessels (Figures 1d, g and j). In β7-integrin staining, large numbers of β7-integrin-positive lymphocytes were seen infiltrating around HEV-like vessels (Figure 1k). In serial section staining, ATX was strongly expressed in these vessels (Figures 1a, c) and was colocalized with CD34+ HEV-like vessels (Figures 1e and h). HEV-like vessels were observed in both CD and UC patients, but not in non-CD/UC patients (Figures 1l–n). We also examined the expression of ATX mRNA in CD patients (n=56). ATX expression levels were significantly higher in inflamed mucosa from CD patients than in the healthy mucosa and quiescent mucosa (Figure 2a). A comparison between two different sites in the same patient showed that ATX expression level in the inflamed area was significantly higher than that in the quiescent area (Figure 2b). We classified samples by endoscopic findings into four groups (redness, erosion, aphtha and ulcer) and compared the degrees of ATX expression. ATX expression was gradually elevated as the degree of mucosal injury became more severe (Figure 2c). The same evaluation was carried out in UC patients (n=159). In the inflamed group, ATX expression levels were significantly higher in the inflamed mucosa from UC patients than in the healthy mucosa and quiescent mucosa (Figure 3a). A comparison between two different sites in the same patient showed that ATX expression levels in the inflamed area were significantly higher than that in the quiescent area (Figure 3b). We classified samples by endoscopic Matts’ grade into four groups (Matts’ 1–4) and compared ATX expression levels in the groups. ATX expression was significantly elevated as endoscopic Matts’ grade became higher (Figure 3c).


Immunofluorescence microscopy of high endothelial venule (HEV)-like vessel. Immunohistochemical study of autotaxin (ATX) from tissues from Crohn’s disease (CD) patient (a) and negative control staining using isotype-matched immunoglobulin G (IgG) staining (b). Higher magnification in CD patients (c and i), and in ulcerative colitis (UC) patients (f) and non-CD/UC patients (l). Immunoreactivity was observed in high-endothelial vessels in CD and UC patients. Immunohistochemical study of CD34 in CD patients (d and j), UC patients (g) and non-CD/UC patients (m). Merged pictures of ATX and CD34 in CD patients (e), UC patients (h) and non-CD/UC patients (n). ATX immnoreactivity was colocalized in CD34 immnoreactivity in CD patients (e) and in UC patients (h), but not in non-CD/UC patients (n). Immunohistochemical study of β7-integrin in CD patients (k). β7-Integrin-positive cell infiltration was observed around HEV-like vessel. The bar represents 50 μm.

Expression of autotaxin (ATX) mRNA in Crohn’s disease (CD) patients. ATX expression levels were considerably higher in inflamed mucosa than that in the healthy mucosa or quiescent mucosa (a). A comparison between two different sites in the same patient showed that ATX expression levels in the inflamed area were considerably higher than that in the quiescent area (b). A comparison among four endoscopic findings (redness, erosion, aphtha and ulcer) showed that ATX expression was gradually elevated as the degree of mucosal injury became more severe (c). The abundance of mRNA was normalized to β-glucuronidase (GUSβ). Differences between two groups were evaluated using the Mann–Whitney U-test. Differences between more than two groups were evaluated using one-way analysis of variance (ANOVA) and a post hoc test. In each test, P<0.05 was considered significant. *P<0.05 as compared with the quiescent group; ¶P<0.05 as compared with the control group; and #P<0.05 as compared with the redness group.

Expression of autotaxin (ATX) mRNA in ulcerative colitis (UC) patients. In the inflamed group, ATX expression levels were considerably higher in the inflamed mucosa from UC patients than in the healthy mucosa and quiescent mucosa (a). A comparison between two different sites in the same patient showed that ATX expression levels in the inflamed area were considerably higher than that in the quiescent area (b). A comparison among four endoscopic Matts’ groups (Matts’ 1–4) showed that ATX expression was considerably elevated as endoscopic Matts’ grade became higher (c). The abundance of mRNA was normalized to β-glucuronidase (GUSβ). Differences between two groups were evaluated using the Mann–Whitney U-test. Differences between more than two groups were evaluated using one-way analysis of variance (ANOVA) and a post hoc test. In each test, P<0.05 was considered significant. *P<0.05 as compared with the quiescent group; ¶P<0.05 as compared with the control group; and P<0.05 as compared with the Matts’ 1 group.
Evaluation of ATX Expression in Intestinal Mucosa of Colitis Model Mice
We examined ATX expression in inflamed colonic mucosa of two different murine colitis models: a CD4+CD25− T-cell transfer model and a DSS model. First, we examined the CD4+CD25− T-cell transfer model mice. The intestinal tracts in these colitis models were compared with those of negative control SCID mice (n=6 per group). Using H–E staining, we observed a significant increase in the number of infiltrating lymphocytes in the inflamed mucosa in the colitis model mice (Figure 4a). Expression of MAdCAM-1 mRNA in the colonic mucosa was significantly increased in the colitis group (Figure 4b). Expression of ATX mRNA in the colonic mucosa was also significantly increased in the colitis group (Figure 4c). Next, we examined the DSS colitis model mice. We treated mice with five cycles of DSS treatment, and we examined the time courses of MAdCAM-1 mRNA expressions, ATX mRNA expression and the number of lymphocytes infiltrating in each cycle of treatment (n=6 per group) (Figures 5a–d). The number of infiltrating lymphocytes gradually increased as the cycles of treatment advanced (Figure 5e). No significant increase in the expression level of MAdCAM-1 mRNA was seen until two cycles of DSS treatment, but the expression level of MAdCAM-1 mRNA increased significantly after three cycles of treatment (Figure 5f). Expression levels of ATX mRNA also increased significantly after three cycles of treatment (Figure 5g), concordant with MAdCAM-1.

Evaluation of inflammation and autotaxin (ATX) in CD4+CD25− T-cell transfer model mouse. To analyze statistics, Mann–Whitney U-test was used and P<0.05 was considered significant. The intestinal tracts in these colitis models were compared with those in severe-combined immunodeficiency (SCID) mice as negative controls. We counted CD3+ cells as lymphocytes (per millimeter muscularis mucosa). The abundance of mRNA was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Histological observations showed a considerable increase in the number of infiltrating lymphocytes in the inflamed mucosa of the colitis model mice (a). Expression of mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1) mRNA in the colonic mucosa was significantly increased in the colitis group (b). Expression of ATX mRNA in the colonic mucosa was also significantly increased in the colitis group (c). *P<0.05 as compared with the quiescent group.

Evaluation of inflammation and autotaxin (ATX) in dextran sodium sulfate (DSS) model mice. (a–d) Representative images of histology. As DSS treatment advanced, inflammatory markers (thickening of colonic wall, cell infiltration and shortening of crypts) became more prominent. The bar represents 100 μm. (e) Number of infiltrating lymphocytes. We counted CD3+ cells as lymphocytes (per millimeter muscularis mucosa). As DSS treatment cycle advanced, the number of infiltrating lymphocytes gradually increased. To analyze statistics, one-way analysis of variance (ANOVA) and a post hoc test were used and P<0.05 was considered significant. *P<0.05 as compared with the cycle 1 group. (f) mucosal vascular addressin cell adhesion molecule 1 (MAdCAM)-1 mRNA expression of DSS-treated mice. The abundance of mRNA was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Expression levels of MAdCAM-1 mRNA were significantly higher in colonic mucosa of chronically developed colitis than in acute colitis. To analyze statistics, one-way ANOVA and a post hoc test were used and P<0.05 was considered significant. *P<0.05 as compared with the cycle 1 group. (g) ATX mRNA expression of DSS-treated mice. The abundance of mRNA was normalized to GAPDH. Expression levels of ATX mRNA were significantly higher in colonic mucosa of chronically developed colitis than in acute colitis. To analyze statistics, one-way ANOVA and a post hoc test were used and P<0.05 was considered significant. *P<0.05 as compared with the cycle 1 group.
Intravital Observation of Lymphocyte Migration to Colonic Microvessels
In the control mice (n=6), the number of T lymphocytes that adhered to and migrated through the colonic mucosal microvessels was very small (Figure 6a). In the CD4+CD25− T-cell transfer model mice (n=6), many T lymphocytes adhered to and migrated through the colonic mucosal microvessels (Figure 6b). However, administration of bithionol to CD4+CD25− T-cell transfer model mice significantly inhibited the transendothelial migration (n=6) (Figures 6c and d). Histological images also showed the prominent inhibitory effect of bithionol on lymphocyte infiltration to colonic mucosa (Figures 6e–h).

(a–c) Distribution of carboxyfluorescein diacetate succinimidyl ester-labeled T lymphocytes in the colonic mucosa of CD4+CD25−-imported mice. In the control mice, the number of T lymphocytes that adhered to and migrated through the colonic mucosal microvessels was very small (a). In the CD4+CD25− T-cell transfer model mice, many T lymphocytes adhered to and migrated through the colonic mucosal microvessels (b). Administration of bithionol to CD4+CD25− T-cell transfer model mice significantly inhibited the transendothelial migration (c) and reduced the number of T lymphocytes transmigrating to outside of the submucosal microvessels (d). The bar represents 100 μm. To analyze statistics, one-way analysis of variance (ANOVA) and a post hoc test were used and P<0.05 was considered significant. *P<0.05 as compared with the bithionol (−) group. (e–h) Representative images of histology in the dextran sodium sulfate (DSS) mouse and the CD4+CD25−-imported mouse. Histological images showed the prominent inhibitory effect of bithionol on lymphocyte infiltration to colonic mucosa. The bar represents 100 μm.
Effects of ATX Inhibition in Colitis Model Mice
We evaluated the inhibitory effects of an ATX inhibitor, bithionol, on colitis activity in CD4+CD25− T-cell transfer model mice and DSS model mice. Each type of model mice was divided into two groups (n=6 per group). One group of mice was infused with bithionol in CMC solution intraperitoneally every other day as an administration group, and the other group of mice was infused with CMC solution intraperitoneally every other day as a control group. CD4+CD25− T-cell transfer model mice were treated for 6 weeks, and DSS colitis model mice were treated for five cycles. The severity of colitis was evaluated by weight-to-length ratio of the colon, histopathology score and number of lymphocytes per high-power field infiltrating into the colonic mucosa. By all of the methods of evaluation, bithionol treatment significantly ameliorated colitis activity in both models of colitis. In gross pathology, treatment with bithionol clearly attenuated thickening of the intestinal wall and shortening of the colon (Figures 7a and 8a). Bithionol treatment also significantly decreased the weight-to-length ratio (Figures 7b and 8b) and the histopathology score (Figures 7c and 8c). The number of lymphocytes infiltrating the colonic mucosa was significantly decreased by bithionol treatment in both models of colitis, but not in either models of healthy control (Figures 7d and 8d).

The inhibitory effect of bithionol on intestinal damage in CD4+CD25−-imported mice. In gross pathology, treatment with bithionol clearly attenuated thickening of the intestinal wall and shortening of the colon (a). Bithionol treatment also significantly decreased weight-to-length ratio (b) and histopathology score (c). The number of lymphocytes infiltrating the colonic mucosa was significantly decreased by bithionol treatment in the colitis groups, but not in either model of healthy controls (d). Differences between two groups were evaluated using the Mann–Whitney U-test. Differences between more than two groups were evaluated using one-way analysis of variance (ANOVA) and a post hoc test. In each test, P<0.05 was considered significant. *P<0.05 as compared with the CD4+CD25−–bithionol (−) group.

The inhibitory effect of bithionol on intestinal damage in dextran sodium sulfate (DSS)-treated mice. In gross pathology, treatment with bithionol clearly attenuated thickening of the intestinal wall and shortening of the colon (a). Bithionol treatment also significantly decreased weight-to-length ratio (b) and histopathology score (c). The number of lymphocytes infiltrating the colonic mucosa was significantly decreased by bithionol treatment in the colitis groups, but not in the healthy control groups (d). Differences between two groups were evaluated using the Mann–Whitney U-test. Differences between more than two groups were evaluated using one-way analysis of variance (ANOVA) and a post hoc test. In each test, P<0.05 was considered significant. *P<0.05 as compared with the DSS–bithionol (−) group.
bEnd3 transendothelial Cell Migration Assay
We examined the effect of the ATX inhibitor on transendothelial migration of lymphocytes by using the bEnd3 cell line. It has been reported that bEnd3 cells show HEV-like morphology after TNFα treatment. First, we examined the expression of ATX mRNA in TNFα-activated bEnd3 cells. TNFα-activated bEnd3 cells showed a significantly higher expression level of MAdCAM-1 mRNA than that in normal bEnd3 cells (Figure 9a). Treatment of TNFα also significantly upregulated ATX expression (Figure 9b). Next, we examined the inhibitory effect of an ATX inhibitor in transendothelial cell migration assays. The number of splenocytes that transmigrated across TNFα-activated bEnd3 cell monolayers was significantly larger than the number that transmigrated across untreated bEnd3 cell monolayers. Bithionol treatment significantly inhibited this increased transendothelial migration by TNFα. BrP-LPA also significantly attenuated the increased transendothelial migration by TNFα, suggesting that transendothelial migration was LPA-dependent (Figure 9c). In immunofluorescent cytometry analyses, the ratio of β7-integrin expression or CD11b expression was not different between splenocytes that migrated through bithionol-treated bEnd3 cell monolayers and splenocytes that migrated through untreated bEnd3 cell monolayers (data not shown), suggesting that the inhibitory effect of bithionol has no tropism for surface expression of adhesion molecules.

Transendothelial cell migration assays using the bEnd3 cell line. Tumor necrosis factor (TNF)α-activated bEnd3 cells showed a significantly higher expression level of mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1) mRNA than normal bEnd3 cells (a). Treatment with TNFα also significantly upregulated autotaxin (ATX) expression (b). In a transendothelial cell migration assay, the number of splenocytes that transmigrated across TNFα-activated bEnd3 cell monolayers was significantly larger than the number that transmigrated across untreated bEnd3 cell monolayers. Bithionol treatment significantly inhibited the increase in transendothelial migration by TNFα. 1-Bromo-3(s)-hydroxy-4-(palmitoyloxy) butylphosphonate (BrP-LPA) also significantly attenuated the increased transendothelial migration by TNFα (c). The abundance of mRNA was normalized to β-glucuronidase (GUSβ). Differences between two groups were evaluated using the Mann–Whitney U-test. Differences between more than two groups were evaluated using one-way analysis of variance (ANOVA) and a post hoc test. In each test, P<0.05 was considered significant. *P<0.05 as compared with TNFα (−) group; #P<0.05 as compared with TNFα (+) bithionol (−) BrP-LPA (−) group.
DISCUSSION
ATX, originally identified as a tumor cell autocrine motility factor, has a major role in extracellular LPA generation through its intrinsic lysophospholipase D (lysoPLD) activity.25 ATX induces cell motility by producing LPA, which acts on LPA receptors.26, 27, 28 ATX is secreted by endothelial cells of HEVs in secondary lymphoid organs such as Peyer’s patches under physiological condition.12 Alternatively, the formation of HEV-like vessels is induced by chronic inflammation in extralymphoid sites, such as the intestinal mucosa of patients with IBD (CD and UC).14, 29 However, whether ATX expression is induced in HEV-like vessels and whether ATX has a role in aberrant lymphocyte migration to inflamed intestinal mucosa remain to be elucidated.
To our knowledge, this is the first evaluation of ATX in IBD patients and murine colitis models. Enhanced expression of ATX mRNA was observed in the inflamed mucosa from both CD and UC patients. ATX expression was colocalized with HEV-like vessels, and it was surrounded by infiltrated lymphocytes. The level of ATX mRNA expression was significantly higher in the actively inflamed mucosa than in the quiescent mucosa in the same patient.
For in vivo studies, we used the CD4+CD25− T-cell transfer and the DSS mouse models of colitis. Adaptive transfer of CD4+CD25− T cells from healthy wild-type mice into SCID mice induces pancolitis and small bowel inflammation at 5–8 weeks after T-cell transfer. Histopathological inspection of distal colons obtained from mice with active disease revealed transmural inflammation, epithelial cell hyperplasia, polymorphonuclear leukocyte and mononuclear leukocyte infiltration, crypt abscesses and epithelial cell erosions. Chronic treatment of BALB/c mice with DSS water also induces pancolitis with extensive crypt and epithelial cell damage, significant infiltration of granulocytes and mononuclear immune cells, tissue edema and ulceration.30
In the CD4+CD25− T-cell transfer colitis model mice, the expression level of ATX mRNA gradually increased as colitis developed. In DSS model mice, expression level of ATX mRNA gradually increased as the number of repetitive DSS treatments increased. This increase was accompanied by an increase in lymphocyte infiltration. These results suggest that ATX is expressed in HEV-like vessels in chronically inflamed colonic mucosa and has an important role in disease progression through aberrant lymphocyte migration.
In addition, we showed the anti-inflammatory effects of an ATX inhibitor, bithionol, in the murine colitis models. Bithionol significantly ameliorated colitis, which was accompanied by a significant decrease in lymphocyte infiltration. Bithionol treatment did not reduce lymphocyte counts in non-inflamed colonic mucosa. In cell migration assays, in contrast to inhibitory effect of the ATX inhibitor on migration to inflamed intestinal colonic mucosal microvessels, the ATX inhibitor did not show any inhibitory effects in non-inflamed intestinal colonic microvessels. In addition, expression of ATX was observed only in inflamed colonic mucosa, but not in non-inflamed colonic mucosa. Previously, it was reported that ATX is involved in lymphocyte migration to high-endothelial vessels of Peyer’s patches of the small intestine, where ATX is abundantly expressed physiologically.11, 12, 13 Collectively, the data suggest that ATX inhibitor does not inhibit lymphocyte migration to colonic mucosal microvessels at steady state.
As we used an ATX inhibitor but not an LPA analog in the colitis models, it is possible that a nonspecific effect of the ATX inhibitor was responsible for ameliorating colitis. However, since measuring the concentration of LPA in tissue is difficult and beyond the capability of our laboratory, we examined the effect of an ATX inhibitor on transmigration of lymphocytes in vitro. A recent study has revealed that bithionol inhibits lysoPLD activity and that it markedly suppresses migration and invasion of melanoma cells.21 Treatment of bEnd3 cells with TNFα induced an HEV-like appearance and ATX expression. The number of lymphocytes that transmigrated through the TNFα-treated endothelium increased as compared with the non-treated endothelium. Both the BrP-LPA treatment and the ATX inhibitor treatment prevented this increase. These findings suggested that the ATX-LPA axis has a role in the transmigration of lymphocytes, not only through physiologically expressed HEV but also through HEV-like vessels that are induced under inflamed conditions.
In transmigration assays, treatment with an ATX inhibitor significantly inhibited lymphocyte transmigration, but surface expression of β7-integrin and CD11b on splenocytes that migrated through epithelial cells was not changed by bithionol treatment (data not shown). This suggests that the role of the ATX–LPA axis in leukocyte transendothelial migration through induced HEV-like vessels has no tropism for surface expression of adhesion molecules on leukocytes. A recent study has shown that the ATX–LPA axis enhances the motility of lymphocytes, and permeability of HEV vessels to accelerate lymphocyte transmigration. It was also shown in that study that ATX does not have a direct relationship with MAdCAM-1.1 Taken together, these findings suggest that the ATX–LPA axis has a role in leukocyte–endothelium interaction, independently of adhesion molecules.
Controlling trafficking is a promising strategy for chronic disease. As the process of lymphocyte migration consists of multiple steps and many molecules are involved in the process, blocking of adhesion molecules alone seems insufficient for satisfactory results. We showed that ATX–LPA has a role in the migration of lymphocytes to the inflamed intestine. Our findings suggest that ATX could be a new target for IBD treatment.
References
Miyasaka M, Tanaka T . Lymphocyte trafficking across high endothelial venules: dogmas and enigmas. Nat Rev Immunol 2004;4:360–370.
von Andrian UH, Mempel TR . Homing and cellular traffic in lymph nodes. Nat Rev Immunol 2003;3:867–878.
Sans M, Panés J, Ardite E et al. VCAM-1 and ICAM-1 mediate leukocyte–endothelial cell adhesion in rat experimental colitis. Gastroenterology 1999;116:874–883.
Briskin M, Winsor-Hines D, Shyjan A et al. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am J Pathol 1997;151:97–110.
Fujimori H, Miura S, Koseki S et al. Intravital observation of adhesion of lamina propria lymphocytes to microvessels of small intestine in mice. Gastroenterology 2002;122:734–744.
Souza HS, Elia CC, Spencer J et al. Expression of lymphocyte–endothelial receptor–ligand pairs, a4b7/MAdCAM-1 and OX40/OX40 ligand in the colon and jejunum of patients with inflammatory bowel disease. Gut 1999;45:856–863.
Subrata G, Eran G, Fiona HG et al. Natalizumab for active Crohn’s disease. N Engl J Med 2003;348:24–32.
Kato S, Hokari R, Matsuzaki K et al. Amelioration of murine experimental colitis by inhibition of mucosal addressin cell adhesion molecule-1. J Pharmacol 2000;295:183–189.
Jesús RN, Timothy O, Giorgos B et al. L-selectin, a4b1, and a4b7 integrins participate in CD4+T cell recruitment to chronically inflamed small intestine. J Immunol 2005;174:2343–2352.
Park EJ, Mora JR, Carman CV et al. Aberrant activation of integrin alpha4beta7 suppresses lymphocyte migration to the gut. J Clin Invest 2007;117:2526–2538.
Kanda H, Newton R, Klein R et al. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat Immunol 2008;9:415–423.
Dietmar V, Martin KW . A new player in lymphocyte homing. Nat. Immunol 2008;9:347–348.
Umemoto E, Hayasaka H, Bai Z et al. Novel regulators of lymphocyte trafficking across high endothelial venules. Crit Rev Immunol 2011;31:147–169.
Jean PG, Timothy AS . High endothelial venules (HEVs): specialized endothelium for lymphocyte migration. Immunol Today 1995;9:449–457.
Palmeri D, Zuo FR, Rosen SD et al. Differential gene expression profile of human tonsil high endothelial cells: implications for lymphocyte trafficking. J Leukoc Biol 2004;75:910–927.
Matts SG . The value of rectal biopsy in the diagnosis of ulcerative colitis. Q J Med 1961;30:393–407.
Okayasu I, Hatakeyama S, Yamada M et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990;98:694–702.
Ostanin DV, Bao J, Kaboziev I et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol 2009;296:G135–G146.
Dieleman LA, Palmen MJ, Akol H et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol 1998;114:385–391.
Miura S, Tsuzuki Y, Fukumura D et al. Intravital demonstration of sequential migration process of lymphocyte subpopulations in rat Peyer’s patches. Gastroenterology 1995;109:1113–1123.
Saunders LP, Ouellette A, Bandle R et al. Identification of small-molecule inhibitors of autotaxin that inhibit melanoma cell migration and invasion. Mol Cancer Ther 2008;7:3352–3362.
Rohnelt RK, Hoch G, Reiss Y et al. Immunosurveillance modelled in vitro: naive and memory T cells spontaneously migrate across unstimulated microvascular endothelium. Int. Immunol 1997;9:435–450.
Jiang G, Xu Y, Fujiwara Y et al. Alpha-substituted phosphonate analogues of lysophosphatidic acid (LPA) selectively inhibit production and action of LPA. Chem Med Chem 2007;2:679–690.
Prestwich GD, Gajewiak J, Zhang H et al. Phosphatase-resistant analogues of lysophosphatidic acid: agonists promote healing, antagonists and autotaxin inhibitors treat cancer. Biochim Biophys Acta 2008;1781:588–594.
Umezu GM, Kishi Y, Taira A et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol 2002;158:227–233.
Gaetano CG, Samadi N, Tomsig JL et al. Inhibition of autotaxin production or activity blocks lysophosphatidylcholine-induced migration of human breast cancer and melanoma cells. Mol Carcinogen 2009;48:801–809.
Khurana S, Tomar A, George SP et al. Autotaxin and lysophosphatidic acid stimulate intestinal cell motility by redistribution of the actin modifying protein villin to the developing lamellipodia. Exp Cell Res 2008;314:530–542.
Pamuklar Z, Federico L, Liu S et al. Autotaxin/lysopholipase D and lysophosphatidic acid regulate murine hemostasis and thrombosis. J Biol Chem 2009;284:7385–7394.
Drayton DL, Liao S, Mounzer RH et al. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol 2006;7:344–353.
Elson CO, Sartor RB, Tennyson GS et al. Experimental models of inflammatory bowel disease. Gastroenterology 1995;109:1344–1367.
Acknowledgements
This research was supported by grants from National Defense Medical College and by Intractable Diseases, the Health and Labor Sciences Research Grants from the Ministry of Health, Labor and Welfare.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Enhanced expression of autotaxin is observed in the inflamed mucosa from Crohn's disease and ulcerative colitis patients. Autotaxin is increased in murine models of colitis, and an autotaxin inhibitor ameliorates colitis through inhibition of lymphocyte migration to colonic mucosa. Autotaxin is therefore a possible target for treatment of these consitions.
Rights and permissions
About this article
Cite this article
Hozumi, H., Hokari, R., Kurihara, C. et al. Involvement of autotaxin/lysophospholipase D expression in intestinal vessels in aggravation of intestinal damage through lymphocyte migration. Lab Invest 93, 508–519 (2013). https://doi.org/10.1038/labinvest.2013.45
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2013.45
Keywords
This article is cited by
-
Autotaxin stimulates LPA2 receptor in macrophages and exacerbates dextran sulfate sodium-induced acute colitis
Journal of Molecular Medicine (2020)
-
Autotaxin-Lysophosphatidic Acid Axis Blockade Improves Inflammation by Regulating Th17 Cell Differentiation in DSS-Induced Chronic Colitis Mice
Inflammation (2019)
-
Dysregulation of lysophosphatidic acids in multiple sclerosis and autoimmune encephalomyelitis
Acta Neuropathologica Communications (2017)
