
Abstract
Hepatocellular carcinoma (HCC) is a very angiogenic and malignant cancer. Conventional chemotherapy is poorly effective because of the abnormal structural organization of HCC-infiltrating vessels. In previous work, we demonstrated that HCC angiogenesis is driven by transforming growth factor beta-1(TGF-β1)/CD105 axis, stimulating liver-derived microvascular endothelial cells (Ld-MECs) migration. As TGF-β1 also affects mural cells (MCs) recruitment and maturation, we asked whether it may contribute to HCC-induced vascular abnormalities. HCC and adjacent non-neoplastic liver (nNL) biopsies obtained from 12 patients were analyzed by immunohistochemistry for angiogenic markers CD105, TGF-β1, CD44 and vascular endothelial growth factor-a (VEGFa) and for MC markers NG2, α-smooth muscle actin (αSMA) and neural cell adhesion molecule (NCAM). The same markers were also investigated by immunocytochemistry on cultured HCC-derived stromal cells (HCC-StCs) and nNL-derived StCs (nNL-StCs) isolated from the same liver biopsies. Angiogenic factors released by StCs were analyzed by ELISA and the interaction between StCs and Ld-MECs by adhesion assay. Compared with nNL, HCC biopsies showed increased angiogenic markers and αSMA that was localized in vessels. By contrast, NG2 and NCAM were substantially localized in tumor cells but absent in vessels and stroma. Cultured HCC-StCs showed less expression of NG2, αSMA and NCAM. They also demonstrated a lower capacity to release angiogenic factors and adhered on Ld-MECs. HCC-StCs and nNL-StCs treated with TGF-β1 or with of HepG2 (a human hepatoma cell line) derived conditioned medium (CM), down-modulated NCAM expression, whereas anti-NCAM antibodies significantly reduced the adhesion of StCs to Ld-MECs. By further blocking TGF-β1 with anti-TGF-β1 antibodies or with Ly-364947 (a specific inhibitor TGF-β1-receptor) adhesion to Ld-MECs and NCAM expression respectively was partially restored. TGF-β1 contributes to HCC-induced vascular alterations by affecting the interaction between HCC-StCs and Ld-MECs through a down-modulation of NCAM expression.
Similar content being viewed by others


Main
A growing tumor recruits new blood vessels to ensure a sufficient quantity of nutrients and oxygen necessary for cancer cell survival and proliferation.1 During this process, neoformed capillaries undergo abnormal maturation2 and vascular abnormalities are often observed in tumor, mainly involving either the composition of the basement membrane or mural cells (MCs) content.3, 4 To this regard, pericytes and smooth muscle cells (SMCs) are fundamental to determine an adequate vessel stability and function.5, 6 Pericytes, in particular, participate in neocapillary construction by producing factors that favor basement membrane deposition7 and endothelial cells (ECs) proliferation.8 By interacting with ECs, they also contribute to the formation of mature vessels.9 Thus, a reduction of MCs in microvessels leads to dilatation, leakage, tortuosity and increased vascular ruptures hallmarks observed in experimental pericyte-deficient mice10 and in tumor vasculature.4 Lack of vessel maturation in tumor determines a hypoxic status, which in turn induces the formation of new vessels.11 Thus paradoxically, solid tumors have increased microvascular density (MVD) but insufficient blood perfusion. Besides the low level of oxygenation, the disorganized architecture of tumor vessels affects several other important pathophysiologic features of tumor: it determines the increment of vascular permeability and interstitial fluid pressure, edema formation and blood stagnation.12 Moreover, even metastatic dissemination and cancer cell invasiveness increase.13
Hepatocellular carcinoma (HCC) is a highly angiogenic, malignant human cancer.14 Its grade of vascularization has been considered an important feature in malignancy and or prognosis.15 In a previous work, we showed that transforming growth factor beta-1 (TGF-β1) and its co-receptors CD105 have an important role in HCC-induced angiogenesis. In particular, we found that TGF-β1 released by HCC, acts as a chemotactic factor for CD105-positive liver-derived microvascular ECs (Ld-MECs).16 However, as TGF-β1 is also involved in vessels maturation through its capacity to recruit MCs and to control their proliferation,17, 18 we asked if the abnormal release of TGF-β1 by HCC may contribute to abnormalities of tumor vessels; by either affecting the content of MCs or by altering their interaction with liver endothelium. To this end, we studied the phenotypic and functional features of liver stroma cells (StCs) both in vivo and in vitro in HCC and in the adjacent non- neoplastic liver (nNL) biopsies. In particular, we investigated the expression of the MC markers NG2 proteoglycan and alpha-smooth muscle actin (αSMA) as well as the neural cell adhesion molecule (NCAM) because of its involvement in pericyte–EC interaction.19 Our findings show, for the first time, that through a down-modulation of NCAM expression, TGF-β1 reduces StCs interaction to Ld-MECs, which may lead to the abnormal morphogenesis of HCC-infiltrating capillaries.
MATERIALS AND METHODS
Patients and Biopsies Characterization
This study involved 12 patients who had undergone curative resection of HCC but had not previously received chemotherapy. This study was approved by the ethical committee board of the Civili Hospital (Brescia, Italy). The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki.
Fresh HCC, adjacent nNL and derma adipose tissues, obtained from the subcute of HCC patients during surgery, were used for StCs isolation, or fixed in 10% formalin buffered and embedded in paraffin for histology sectioning and immunoperoxidase studies. HCC biopsies were also stained with hematoxylin–eosin for routine histological evaluation of malignancy grade, determined according to Edmodson classification20 that allowed the selection of patients with a similar high (G2) grade for this study. The active rate of cell proliferation was calculated by using Ki67 (DakoCytomation, Glostrup, Denmark), whereas MVD was measured according to a previously described procedure.21 Additional features, such as tumor size, venous invasion and secondary microsatellite lesions, were also investigated.
Histology and Immunohistochemical Studies
HCC and nNL sections were immunostained for proteins of interest according to standard protocols.16 Briefly, sections were transferred to glass slides coated with poly-L-ysine, deparaffinized in 100% xylene and rehydrated in graded ethanol. After heat-induced antigen retrieval, endogenous peroxidase was inhibited by incubating tissue sections with 3% hydrogen peroxidase for 15′ at room temperature, whereas a specific epitope binding was blocked by incubation for 20′ with 20% human serum. All samples were then processed by the avidin–biotin peroxidase complex method according to the manufacturer's recommendations (LASB kit, DakoCytomation). The antibodies used for this study were anti-CD105, anti-NG2, anti-CD14 (dilution 1:50), anti-TGF-β1 (dilution 1:200) and anti-Ve-cadherin (Ve-cad; dilution 1:30) purchased from Santa Cruz Biotechnology (CA, USA); anti-β catenin (β-cat; dilution 1:5) and anti-E-cadherin (E-cad; dilution 1:50) purchased from Zymed Laboratories (San Francisco, CA, USA); anti-NCAM (CD56; dilution 1:50), anti-vascular endothelial growth factor a (VEGFa; dilution 1:100), and anti-CD49d/VLA-4 integrin (dilution 1:10) and anti-CD34 (dilution 1:30) purchased from NeoMarkers (Fremont, CA, USA); anti-αSMA (dilution 1:400, DakoCytomation) and anti-CD44 (dilution 1:30; Novocastra Laboratories, Newcastle, UK). A quantitative analysis of NCAM, NG2 and αSMA expression, in nNL and in HCC specimens, was also preformed according to a procedure used for the evaluation of MVD. Three different zones: parenchyma, stroma and vessels of the histological sections were selected for counting the positive cells for each MC marker.
Anti-CD44, anti-CD105, anti-TGF-β1, anti-NCAM, anti-NG2, anti-CD49d/VLA-4, anti-αSMA and Ki67 antibodies were also used for immunocytochemical studies performed on cultured StCs by utilizing the avidin–biotin peroxidase complex described above. A quantitative evaluation of the expression of MC and mesenchymal markers was investigated after avidin–biotin peroxide staining. Positive cells for each marker tested were counted at × 40 magnification, calculating the percentage of positive cells of total cells counted in each field.
Analysis of MCs in HCC and nNL Biopsies by Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
The expression of ACTA2/αSMA, CD34, CSPG4/NG2, NCAM1, TGF-β1 and VEGFa mRNAs from normal and tumoral samples, stored in liquid nitrogen, were analyzed by a real-time RT-PCR assay using the 7500 Fast Real-Time PCR System (Applied Biosystems, Life Technologies Europe BV, Bleiswijk, Netherlands). Total RNA was extracted using NucleoSpin RNA II kit (Macherey-Nagel GmbH KG, Düren, Germany) and treated with DNase. The cDNAs were synthesized by random hexamers and Taqman reverse transcription reagents (Applied Biosystems) from 800 ng of total RNA, in a total volume of 25 μl, 1.5 μl of the cDNA synthesis reaction was used for the specific amplification of the target transcript and of the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase, as normalization control.
Primers and probes were bought from Applied Biosystems (CD34: Hs00156373_m1, VEGFα: Hs00900055_m1, TGF-β1: Hs00998133_m1, NCAM1: Hs00941821_m1, ACTA2: Hs00909449_m1, CSPG4: Hs00426981_m1) or synthesized according to the Applied Biosystems recommendations. The threshold cycle (CT) was determined for each sample in duplicates and quantification was performed using the comparative CT method (ΔΔCT).
Isolation and Culture of StCs
HCC, nNL and adipose-derived StCs (HCC-StCs, nNL-StCs and Ad-StCs, respectively) were isolated and cultured after minor modifications of a previously described procedure.16, 22 Briefly, specimens were finely minced with scissors; HCC and nNL were digested for 2–3 h at 37 °C in RPMI-1640 (Sigma, St Louis, MO, USA) containing 0.2% of collagenase type II (Sigma), whereas adipose tissue was incubated with collagenase plus 1:100 dilution of DNAase (Sigma) and washed with PBS. The pellet of cells was resuspended in RPMI-FCS and filtered (100 μm pore size) to remove undigested tissue. Cells were plated on T25 flasks with RPMI-1640 supplemented with 10% FCS (RPMI-FCS) and left to adhere for 48 h; then, floating cells were discarded by aspiration, whereas adherent cells were fed with fresh RPMI-FCS. After 4–5 days, the adherent cells showing a fibroblastic-like morphology (as confirmed by microscope examination) were detached by trypsinization and seeded to expand in a new T25 flask. This was considered the first passage. The resulting HCC-StCs, nNL-StCs and Ad-StCs cultures were maintained in RPMI-FCS and were passed weekly at 1:2 splitting ratio. The growth rate of the three types of StCs was assessed by counting the cells with hemocytometer. For the experimental procedures, StCs were used within 4 to 6 passages.
Evaluation of Angiogenic Factors Released by StCs
Growth factors and cytokines released by HCC-StCs, nNL-StCs and Ad-StCs in the culture medium were detected by ELISA. Two ml of conditioned medium (CM) from HCC-StCs (HCC-StCs-CM), nNL-StCs (nNL-StCs-CM) and Ad-StCs (Ad-StCs-CM) cultures were collected after 72 h, at 70–80% of cell confluence and between 3 and 5 passages. Afterward, cells were detached with trypsin, stained with Trypan blue to detect cell viability and counted in order to normalize the amount of released angiogenic factors in respect to the number of cells. VEGFa, TGF-β1, hepatocyte growth factor (HGF), angiopoietin-1 (Ang-1), and 2 (Ang-2) were quantified with Elisa kits (R&D System UK), according to the manufacturer's instructions. The specific protein concentration in CMs was detected in accordance with the standard guideline protocol supplied with ELISA kits and by subtracting the amount of each angiogenic factor contained in control medium (CTRL). Absorbance was measured at 450 nm with microplates photometric reader DV990BV4 (GVD, Milan, Italy). Data were expressed as mean±s.d. of the secreted factor per 106 cells. Assays were repeated twice and each sample was run in triplicate. Following the same procedure, we also analyzed the amount of TGF-β1 present in the CM obtained from culture of human HCC cell line (Hep G2) (purchased from Centro Substrati Cellulari, IZSLER, Brescia, Italy). This line was cultured in RPMI medium supplemented with 10% FCS, 10 units/ml penicillin and streptomycin (Euroclone, Milan, Italy) and 2 mM glutamine (Invitrogen and Life Technologies, NY, USA) and was passed twice a week at split ratio 1:5. HepG2-derived CM (HepG2-CM) was obtained by culturing 106 cells for different intervals of time (24, 48 and 72 h). At the end of each incubation time, the culture medium of HepG2 was aspirated, centrifuged and stored at −30 °C before use.
StCs Adhesion to ECs
Adhesion experiments of StCs to ECs were performed following a previously described procedure.23 Briefly, primary cultures of Ld-MECs obtained as previously described,16 and human umbilical vein ECs (HUVECs), purchased from Lonza (Verviers, Belgium), were plated onto collagen-coated 24-multiwell plates at a concentration of 5 × 104/500 μl in complete endothelial basal medium (EGM Bullet kit, Lonza). The plates were incubated for 4 to 5 days to obtain a cell monolayer. Both Ld-MECs and HUVECs were activated by adding tumor necrosis factor-α (TNFα; 25 ng/ml; Sigma Chemical) for 12 h. In some experiments, ECs were also treated for 24 h with TGF-β1 (50 ng/ml; Sigma) alone or combined with TNFα. ECs were then washed with PBS and allowed to interact with the different cultures of StCs (104 cells per well) in RPMI-1640 containing 0.2% bovine serum albumin (BSA). Adhesion experiments were also performed with StCs primed for 24 h with TGF-β1 (50 ng/ml) alone or combined anti-TGF-β1 (1 μg/ml) blocking monoclonal antibodies (R&D System, Minneapolis MN, USA) and with TNFα (25 ng/ml). Alternatively, StCs were primed with anti-NCAM or with anti-CD49d/VLA-4 (diluted 1:10 and 1:50 in RPMI-1640+0.2% BSA, respectively) by incubating for 6 h at 37 °C under gentle agitation, then StCs were seeded on ECs and incubated for further 30′ at 37 °C. The unbound StCs were removed by three washes with warm PBS and StCs attached to ECs were fixed for 5′ with 100 μl of cold methanol and stained with Diff Quick (Merz-Dade, Dudingen, Switzerland) for 30′ at room temperature. The plates were then washed several times with deionized water and the StCs bound to ECs were counted with a calibrated eyepiece in 15 different fields at × 40 magnification. Each test was run in quadruplicate.
Angiogenic Potential of StCs
To assess the angiogenic potential of StCs, CMs, collected on 72 h of culture of HCC-StCs, nNL-StCs and Ad-StCs were added (at different dilutions) to Ld-MECs and HUVECs. Proliferation assay was performed as previously described16 and the proliferative activity induced by each HCC-StCs-CM preparation was compared with that of the corresponding nNL-StCs-CM prepared from the same patient and to Ad-StCs-CM used as a referee for normal StCs strain.
Matrigel (Sigma) assay was used24 to test the effect of the different StCs on vascular morphogenesis. Briefly, 50 μl of Matrigel were seeded into cold wells of a 96-multiwell plate. After Matrigel jellification at 37 °C for 30′, Ld-MECs and HUVECs were seeded on Matrigel at concentration of 104 cells/ml in 50 μl of endothelial basal medium (diluted 1:2) containing HCC-StCs-CM, nNL-StCs-CM and Ad-StCs-CM. In some experiments, we also add anti-Tie2 (5 μg/ml) polyclonal antibodies (R&D System) to block the activity of Ang-1 present in the HCC-StCs-CM.
After 4 and 7 days, the number of cords were counted using inverted microscopy at × 10 magnifications. Assays were repeated twice and each sample was run in triplicate.
Flow Cytometry Analysis for Adhesion Molecules (AMs) Expression
AMs expression by HCC-StCs, nNL-StCs and Ad-StCs was analyzed by flow cytometry (FC) before and after TGF-β1 stimulation. Briefly, cells were washed and part of them was incubated for 24 h with 50 ng/ml of TGF-β1. Then they were washed again, trypsinized, resuspended with FC buffer (PBS BSA 0.5%, sodium azide 0.02%, pH 7.2) and fixed with paraformaldehyde 2%. Cells (1 × 105/500 μl) were incubated for 20′ at room temperature, in the dark, with CD56/NCAM-phycoerythrin (Serotech, Italy), CD44- and CD54/ICAM-1-fluorescein isothiocynate (Immunotech, Milan, Italy) and CD49d/VLA-4 (Biolegend, Italy) antibodies, and then washed with FC buffer. In all, 20 000 events were acquired for each analysis using Epics ‘XL-MLC’ (Beckman Coulter, USA) and histogram elaboration was performed with EXPO 32 software. Alternatively, we also investigated the capacity of HepG2 to affect NCAM expression on HCC-StCs, nNL-StCs and Ad-StCs. Specifically, StCs were cultured for 12 days in the presence of HepG2-CM mixed 1:1 with the RPMI-FCS control culture medium. In some experiments, we added to HepG2-CM, Ly-364947 a compound that selectively inhibits TGF-β type I receptor kinase activity25 (TGF-βRI, ALK5) (Sigma, MI, USA) at the final concentration of 59 nM.
RPMI-FCS control medium and HepG2-CM with or without Ly-364947 were refreshed every 2 days for a total of five times. At the end of 12 days of culture, the StCs were detached by trypsin and the expression of CD56/NCAM were analyzed as described above.
Statistical Analysis
Results are expressed as mean±s.d. Statistical significance was evaluated by analysis of variance followed by Tukey–Kramer multiple comparison test and by Student's t-test.
Statistical significance of differences was set at P-value <0.05.
RESULTS
Different Pattern of Expression of NG2, αSMA and NCAM in HCC Biopsies
Most of the HCC patients had a cancer nodule >2 cm in diameter, tumor vascular invasion, abnormal capillary architecture and increased MVD. The pathological features of their biopsies were reported in the Supplementary Table 1. The analysis of the in situ expression of angiogenic and cell adhesion markers confirmed previous results16 demonstrating that, compared with the adjacent nNL tissues, all G2 biopsies showed a different pattern of Ve-Cad, β-cat and CD14 expression and an extremely high level of angiogenic markers such as CD34, CD105, CD44, TGF-β1 and VEGFa (Supplementary Figure 1). The presence of pericytes and SMCs, detected by the expression of NG2 and αSMA, respectively,26, 27 and NCAM, an AM involved in EC–pericyte interaction,19 was significantly different in HCC compared with their nNL counterpart. In HCC, NG2 and NCAM stained intensively neoplastic hepatocytes (Figures 1a and g) and were almost absent in HCC stroma (Figures 1c and i) and in tumor vessels (Figures 1e and k). On the contrary, NG2 and NCAM were lower in hepatocytes of nNL (Figures 1b and h), but present in stroma (Figures 1d and j) and vessels (Figures 1f and l). The αSMA was significantly more highly expressed in both small arterioles and sinusoids of HCC (Figures 1n, p and r). In nNL, αSMA labeled specifically only small arterioles but not sinusoids (Figures 1m, o and q). HCC also showed an intense and diffuse capillarity, with many capillaries staining intensively for αSMA.

Localization and expression of NG2, αSMA and NCAM in HCC and in nNL biopsies. The localization of the pericyte marker NG2 (panels a–f), NCAM (panels g–l) and SMC marker αSMA (panels m–r) were investigated. NG2 stained intensively several neoplastic hepatocytes (panel a), it was absent in HCC stroma (panel c) and in vessels (panel e). By contrast, NG2 was less present in the parenchyma of nNL (panel b) but significantly present in stroma (panel d) and in vessels (panel f). Similarly, NCAM was present in neoplastic hepatocytes (panel g) absent in HCC stroma (panel i) and vessels (panel k), less expressed in nNL parenchyma (panel h) but vice versa, present in nNL stroma (panel j) and vessels (panel l). In nNL, αSMA stained only arterioles but not sinusoid (panels m and o) even in the presence of inflammatory foci (panel q); in HCC, αSMA stained intensively both arterioles and sinusoids (n and p). Several αSMA-positive cells lining the capillary lumen (panel r), at higher magnification, seem of epithelial-like morphology suggesting a neoplastic origin (square insert in panel r). Black arrow and white arrows in panel g indicate neoplastic hepatocytes and nerve fibers staining positively for NCAM, respectively. The positive staining of nerve fibers for NCAM indicates the specificity of anti-NCAM antibodies used. In the square insert (magnification × 20) in panel r, the white arrow indicates round cells positive to αSMA. Photos were taken at magnification × 10 after avidin–biotin peroxide staining.
Interestingly, αSMA+ cells lining capillary lumen showed an epithelial-like morphology with a rounded shape similar to that of neoplastic hepatocytes (Figure 1r, square insert). The expression of NG2, NCAM and αSMA in parenchyma, stroma and vessels, for each HCC and their counterpart nNL specimens, was quantified by counting the positive cells after staining (Supplementary Table 2). Compared with nNL, the results confirmed an altered pattern of expression of MC markers in HCC. A substantial increment of αSMA, and a significant reduction NCAM was observed, whereas NG2 expression increased significantly but only in HCC parenchyma. The expression of MCs (ACTA2/αSMA, CSPG4/NG2 and NCAM) and some angiogenic (CD34, TGF-β1 and VEGFα) markers, was also investigated by RT-PCR. As shown in the Supplementary Figure 2, the mRNA quantification of each marker analyzed, confirmed the trend observed by immunohistochemistry: the angiogenic markers CD34, TGF-β1 and VEGFα were increased in HCC, as well the MC markers NG2 and αSMA, whereas NCAM was significantly down-modulated.
Decrease in Mesenchymal and MC Markers Expression on Cultured HCC-StCs
Successful isolation and expansion of both HCC-StCs and nNL-StCs counterpart were obtained from 10 of the 12 patients. The number of StCs varied among specimens and rarely exceeded 20–30% of the total seeded cells. In some cases (1/3 out of 12), the nNL-StCs showed a higher growth rate index and were easier to expand in vitro than HCC-StCs obtained from the same patient (Supplementary Table 3). Ad-StCs were also easy to culture and had a good proliferative capacity (data not shown). Labeling studies demonstrated that nNL-StCs were positive for NG2, αSMA, NCAM, CD44, CD105, TGF-β1 and CD49d/VLA-4, although their staining was significantly less intense than in Ad-StCs (Figure 2). By contrast, cultured HCC-StCs were only slightly positive for CD44, CD105, αSMA, TGF-β1, NCAM and CD49d/VLA-4 and were negative for NG2. The lack of Ki67 staining confirmed that HCC-StCs had a lower proliferative activity in vitro. A quantification of MC and mesenchymal markers expression was obtained by counting the positive cells to avidin–biotin peroxide staining.

Expression of MC and mesenchymal markers on cultured StCs. Ad-StCs were strongly positive for αSMA, NCAM, TGF-β1, CD44 and CD105, less for NG2 and the CD49d/VLA-4 integrin. Similarly, nNL-StCs were positive for all markers but significantly less than Ad-StCs. By contrast, HCC-StCs were only slightly positive for αSMA, NCAM, TGF-β1, CD44, CD105 and CD49d and almost negative for NG2. Ki67 positivity indicates cells undergoing division. Pictures of cultured StCs in the figure are representative of the total 10 investigated. Photos were taken at magnification × 10 after avidin–biotin peroxide staining.
Compared with cultured HCC-StCs, nNL-StCs have a significantly higher percentage of cells positive for NG2, NCAM, TGF-β1, CD49d /VLA-4 and αSMA (Supplementary Table 4).
HCC-StCs Display a Reduced Angiogenic Potential and Capacity to Induce Vascular Morphogenesis
With few exceptions, CMs collected from cultures of nNL-StCs always contained a higher level of HGF, VEGFa, TGF-β1, Ang-1 and Ang-2 angiogenic factors compared with CMs collected from their HCC-StCs counterparts (Table 1). In spite of a high variability among cultures, the mean of VEGFa and Ang-1 production was significantly higher in nNL-StCs. Moreover, all nNL-StCs-CM-contained detectable quantities of both Ang-1 and Ang-2, which were detectable only in 6/10 and in 7/10 HCC-StCs-CM preparations, respectively (Table 1). The production of angiogenic factors by both nNL-StCs and HCC-StCs was significantly lower if compared with that of Ad-StCs of the same patients (Supplementary Figure 3). The addition of nNL-StCs-CM to both HUVECs and Ld-MECs was more effective in improving proliferation than the addition of HCC-StCs-CM and, in some patients, there was a correlation between the concentration of the VEGFa in the CM and the capacity to stimulate ECs proliferation (Supplementary Figure 4). We then evaluated if HCC-StCs and nNL-StCs may affect vascular morphogenesis by culturing ECs on Matrigel.24 As shown in Figure 3, the average value of cords formed in the presence of HCC-StCs-CM was significantly lower compared with those observed with nNL-StCs-CM and CTRL medium. Ad-StCs-CM particularly improved the number of cords formed by Ld-MECs (Figure 3a). Interestingly, the lowest number of EC cords found was after the addition of HCC-StCs-CM derived from patient 8 (Figure 3b), in which Angs were not detected (Table 1). This underlines the importance of Angs in vascular morphogenesis induced by Matrigel.28, 29 To verify if this was the case, we evaluated the cord formation under the presence of nNL-StCs-CM and HCC-StCs-CM derived from patient 9 that contained a good amount of Ang-1 and 2 (Table 1). In addition, we also added antibodies anti-Tie2 that block Ang-1 receptor activity.30 As shown in Figure 3b, HCC-StCs-CM and nNL-StCs-CM of patient 9 produced a good number of cord formations closely similar to the control medium that was significantly reduced by anti-Tie2 antibodies. Finally, cords regression that started after 3 days in cultures grown in CTRL medium, was not accelerated by the addition of HCC-StCs-CM; however, the addition of nNL-StCs-CM and, in particular, Ad-StCs-CM delayed cords regression to day 4 (Figure 3c).

Effect of CM derived from cultured StCs on Ld-MECs morphogenesis induced by Matrigel matrix. (a) Cords either formed by HUVECs or Ld-MECs under the influence of HCC-StCs-CM, nNL-StCs-CM and Ad-StCs-CM (all CMs were used at 1:1 dilution with control medium). (b) The effect on cord formations by HCC-StCs-CM from patient #8 and 9. In these CMs, by ELISA assay, no Angs were detected in patient #8, whereas patient #9 showed a significant presence of Ang 1, in both nNL-StCs-CM and HCC-StCs-CM. Anti-Tie2 mAbs were also used (at 5 μg/ml) to block Ang-1-dependent cord formations. (c) Photographs (magnification × 10) taken on day 4 when cords formed by Ld-MEC underwent regression. Note that cords regression was delayed by nNL-StCs-CM and Ad-StCs-CM but was not affected by HCC-StCs-CM addition (at 1:1 dilution). Further, the addition of anti-Tie2 significantly blocked cord formations in patient #9. Columns in the figure are number of cords counted/field expressed as mean±s.d. and are the average of results obtained by testing 10 different preparations for each StCs strain. *P<0.05, **P<0.01 versus nNL-StCs-CM ; °P< 0.05 versus, °°P< 0.01 versus no anti-Tie2 addition.
Reduced Capacity of HCC-StCs to Adhere on Ld-MECs is Improved by TGF-β1
Adhesion of HCC-StCs on quiescent Ld-MECs was lower than that of nNL-StCs counterpart, but both were less adhesive if compared with Ad-StCs (Figure 4a). Activation of Ld-MECs with TNFα improved nNL-StCs and Ad-StCs adhesion more than that of HCC-StCs and amplified the differences between HCC-StCs and nNL-StCs (Figure 4a). Priming of Ld-MECs with TGF-β1 alone or in combination with TNFα (25 ng/ml) did not change the StCs adhesion levels (Figure 4b). By contrast, the priming of StCs with TGF-β1, while slightly reducing adhesiveness to quiescent Ld-MECs monolayer, induced a potent inhibition of StCs adhesion on TNFα-activated Ld-MECs (Figure 4c). Under stimulation of Ld-MECs with TNFα, the inhibition of StCs adhesion induced by TGF-β1 was antagonized by the presence of anti-TGF-β1 antibodies as shown in Figure 4c. The priming of StCs with TNFα did not affect their adhesion either on quiescent or TNFα-activated Ld-MECs (Figure 4d).

Adhesion of HCC-StCs, nNL-StCs and Ad-StCs to Ld-MECs. (a) StCs detached from culture were seeded on ‘quiescent’ or TNFα-activated Ld-MEC monolayer. Note that HCC-StCs adhere significantly less than nNL-StCs counterpart particularly to TNFα-activated Ld-MECs (*P< 0.05 versus nNL-StCs; °P<0.05, °°P<0.01 versus quiescent Ld-MECs). (b) Priming of Ld-MECs with TGF-β1 did not affect basal adhesion of StCs even if combined with TNF-α. (c) StCs primed with TGF-β1 (50 ng/ml for 24 h) significantly reduced adhesion of all different StCs strains when seeded on TNFα-activated Ld-MECs (*P<0.05, **P<0.01 versus quiescent Ld-MECs). (d) Priming of StCs with TNFα ( 25 ng/ml for 24 h) did not affect adhesion of StCs to quiescent and TNFα-activated Ld-MECs (*P<0.05 versus nNL-StCs; °P<0.05, °°P<0.01 versus quiescent Ld-MECs). All the columns in the figure are the number of StCs counted in each field ( × 40 magnifications) expressed as mean±s.d. and are the average of results obtained by testing 10 different preparation of StCs for each strain.
TGF-β1 Down-Modulate NCAM Expression on StCs
In order to verify whether TGF-β1 affects StCs adhesion through a mechanism involving AMs expression, AM expression was analyzed by FC after exposure of StCs to TGF-β1 for 24 h. The analysis confirmed the immunocytochemical results showing that, under basal conditions, HCC-StCs had a lower expression of NCAM, CD44 and the CD49d/VLA-4 integrin, with only CD54/ICAM-1 similarly expressed on HCC-StCs and nNL-StCs (Figure 5). After priming with TGF-β1, StCs showed a significant down-modulation of NCAM (Figure 5a), whereas the expression of CD44 (Figure 5b), CD54/ICAM-1 (Figure 5c) and CD49d/VLA-4 (Figure 5d) were not significantly affected. Similarly, incubation of StCs with TNFα alone or in association with TGF-β1 did not significantly change AMs expression, although a slight increment of CD54/ICAM-1 was noted (data not shown).

AMs and CD49d/VLA-4 integrin expression of HCC-StCs, nNL-StCs and Ad-StCs after priming with TGFβ1. (a) NCAM, (b) CD44, (c) CD54/ICAM-1 and (d) CD49d/VLA-4. Note that, TGF-β1 reduces significantly NCAM but do not affect CD44, CD54/ICAM-1 and CD49d/VLA-4 expression. Values are the mean±s.d. of independent experiments performed with 10 different preparation of StCs for each strain. *P<0.05, **P<0.01 versus nNL-StCs; °P<0.05, °°P<0.01 versus not TGF-β1-treated StCs.
Human Hepatoma Cell Line HepG2 release TGF-β1 and its CM Induced Down-Modulation of NCAM Expression on HCC-StCs and nNL-StCs
As HCC in vivo express and produce high level of TGF-β1,15 we asked whether the chronic exposure of StCs to hepatoma cells may affect NCAM expression. To this end. we cultured the human hepatoma cell line HepG2, which was previously shown to express multiple cytokines genes including TGF-β1,31 in order to obtain significant quantity of CM. As shown in the Supplementary Figure 5, we found that HepG2 secreted good quantities of TGF-β1 in the culture medium, around 200 pg/ml during the first 24 h, that increased to around 1200 pg/ml after 72 h of culture. Afterward, we cultured for 12 days HCC-StCs, nNL-StCs and Ad-StCs in the presence HepG2-CM diluted 1:1 with control medium. In some experiments, we also added an inhibitor of TGF-β1, the compound Ly-394947 that is shown to selectively inhibit TGF-β1 receptor kinase activity.25 We found that the chronic treatment of StCs with HepG2-CM induced significant reduction of NCAM expression, in particular on HCC-StCs and nNL-StCs, but did not reduce NCAM on Ad-StCs (Figure 6). Interestingly, the addition of Ly-394947, to block TGF-β1 activity in HepG2-CM, did restore NCAM expression on both HCC-StCs and nNL-StCs, but not at the level of untreated cells. By contrast, Ly-394947 was able to antagonize almost completely the down-modulation of NCAM produced by the TGF-β1 at optimal concentration (Figure 6).

Expression of NCAM on StCs under stimulation of HepG2-CM and in the presence of Ly-364947 a TGF-β1 inhibitor. HCC-StCs, nNL-StCs and Ad-StCs were cultured for 12 days in the presence HepG2-CM diluted 1:1 with control medium and with/out the addition of Ly-394947 (59 nM) a selective inhibitor of TFG-β1 activity. At the end of culture, NCAM expression was analyzed by FC. Note that HepG2-CM induced a significant NCAM down-modulation particularly on HCC-StCs and nNL-StCs and was stronger than those obtained with 50 ng/ml of TGF-β1 (positive control). Interestingly, the addition of Ly-394947 to HepG2-CM only partially restored NCAM expression on StCs. Columns are the mean±s.d. of independent experiments performed with five different preparation of StCs for each strain. *P<0.05, **P<0.01 versus NCAM expression on HCC-StCs, nNL-StCs and Ad-StCs cultured in CTRL (control medium).
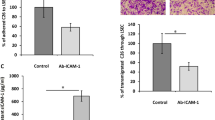
Anti-NCAM and CD49d/VLA-4 Antibodies Reduce Adhesion of StCs to Ld-MECs
To investigate whether the reduced capacity of HCC-StCs to adhere to Ld-MECs could be a consequence of a lower expression of NCAM, we performed adhesion experiments after priming StCs with anti-NCAM, and using anti-CD49d/VLA-4 integrin to block adhesion on activated ECs.32 As shown in Figure 7, both anti-NCAM and anti-CD49d/VLA-4 reduced the adhesion of StCs. More specifically, anti-NCAM reduced the adhesion of HCC-StCs, nNL-StCs and Ad-StCs on quiescent Ld-MECs by 60%, 45% and 43% and by 72%, 56% and 55% on TNFα-activated Ld-MECs, respectively. As expected, adhesion of StCs to ECs was highly inhibited by anti-CD49d/VLA-4.

Effect of anti-NCAM and anti-CD49d/VLA-4 on HCC-StCs, nNL-StCs and Ad-StCs adhesion on Ld-MECs. The adhesion of StCs to quiescent and TNFα-activated Ld-MECs was evaluated after priming with anti-NCAM and CD49d/VLA-4 antibodies. In the figure, photographs of adherent StCs, that appears as round cells on the top of Ld-MECs monolayer. (a) Quiescent Ld-MECs monolayer. (b) TNFα-activated Ld-MECs monolayer. Note that anti-NCAM was able to reduce adhesion of HCC-StCs, nNL-StCs and Ad-StCs as well as anti-CD49d/VLA-4 antibodies that were used as positive control for inhibition of StCs adhesion. Photographs in the figure are representative of 10 different preparations for each kind of StCs investigated. On the left corner, upper part, of each photograph the number of StCs±s.d. that adhered to Ld-MECs is reported and counted at × 20 magnifications.
DISCUSSION
Tumor vessel abnormalities are probably one of the major causes in determining the scant efficacy of chemotherapy in HCC patients. Previous data have shown that TGF-β1 released by cancer cells seems to have a fundamental role in HCC progression and angiogenesis.16, 33 However, because TGF-β1 is involved in vascular maturation,19, 20 in this study, we asked whether TGF-β1 could affect the morphogenesis of HCC-infiltrating vessels. To this end, we studied the angiogenic properties, the expression of MC markers NG2 and αSMA, and AMs, in particular NCAM, because of its involvement in pericyte–EC interaction9, 19 in HCC-StCs and nNL-StCs. These cells were successfully cultured from HCC, adjacent nNL and adipose tissue of patients with high grade HCC malignancy. In two patients, we failed to expand HCC-StCs and this result, together with a lower proliferative index and expression of Ki67, demonstrated that HCC-StCs were less prone to expand in culture than their nNL-StCs counterpart.
Using this approach, we demonstrate, for the first time, that TGF-β1 reduces the interaction between HCC-StCs and liver ECs through its capacity to down-modulate NCAM expression, and thus affect the maturation of HCC-infiltrating vessels. In addition, to provide a mechanistic explanation of how TGF-β1 induces vascular abnormalities, our study highlights some important new features of HCC-infiltrating StCs.
First of all, although NG2 was significantly expressed in neoplastic hepatocytes, the finding that HCC stroma displayed a low expression of NG2 and a significant increment of αSMA compared with nNL, suggests that an abnormal MCs composition rather than a lower MCs content, affected HCC-infiltrating vessels.4, 11 In particular, the reduced number of pericytes, evidenced by the low expression of NG2 in vessels, could explain why HCC vessels leak, whereas an increment of αSMA+SMCs in vessel walls may be responsible of the morphological abnormalities that are typically described in tumor vessels of poorly differentiated HCC.34, 35 Interestingly, cultured HCC-StCs showed a low expression of αSMA. A possible explanation of the discrepancy with in vivo data is that cells lining the vessel lumen that abnormally express αSMA might be neoplastic hepatocytes, as suggested by their epithelial-like shape. Therefore, the wall of HCC-infiltrating capillaries could be composed of MCs of both normal and neoplastic origin.
Our study also demonstrated a significant alteration of NCAM expression both in HCC biopsies and cultured HCC-StCs. NCAM expression has been implicated in the progression of different human cancers, being downregulated in some cancers36, 37 and upregulated in others.38, 39 The expression of NCAM in hepatic cancers has been poorly investigated but, initially, this marker has not been associated to HCC and cholangiocarcinoma.40 More recently, NCAM was found in cancer-initiating stem cells of the liver.41, 42 We demonstrated that in HCC, NCAM, although present in several cancer cells, was significantly down-modulated and practically absent in stroma and in vessels, thus indicating that, contrarily in other cancers,43, 44 NCAM does not stain tumor vessels of HCC.
The mechanism of how NCAM may regulate pericyte–EC interaction is not well understood, particularly in tumors, but the absence of NCAM in tumor stroma seems less important for inducing vascular defects than its expression on tumor cells.45 Our in vitro studies on HCC-StCs strongly support the contrary. Indeed, we found that, compared with nNL-StCs counterpart and to Ad-StCs, HCC-StCs had a lower basal expression of NCAM and this correlated with a reduced capacity to adhere on Ld-MECs. In addition, TGF-β1 down-modulated NCAM expression on StCs, reducing their adhesion to Ld-MECs. An action that was restored by blocking TGF-β1 with anti-TGF-β1 antibodies. Furthermore, we found that anti-NCAM antibodies inhibited the adhesion of StCs to Ld-MECs as well as anti-CD49d/VLA-4 antibodies, which blocks the binding to vascular cell AM-1 on activated ECs.46 All together, these data indicate that HCC-infiltrating StCs have less capacity to interact with liver vessels because of reduced NCAM expression. This could be caused by the continuous and abnormal release of TGF-β1 by HCC cells.33 Indeed, our data support this hypothesis because we found that in vitro the chronic exposure of HCC-StCs and nNL-StCs to HepG2, a human hepatoma cell line that releases significant amounts of TGF-β1, reduced NCAM expression which, in turn, was partially restored by Ly-364947, a compound described as selectively inhibiting TGF-β type I receptor kinase activity.25 However, it is not clear how TGF-β1 down-modulate NCAM expression on StCs. It is shown that NCAM and E-cadherin have a key role in epithelial to mesenchymal transition in cancer.47 TGF-β represses E-cadherin expression through Smad-mediated transcription and enhances NCAM expression on cancer cells that leads to form metastasis.48, 49
Focusing on HCC, we found that neoplastic hepatocytes, particularly at tumor periphery, expressed NCAM and were strongly positive for TGF-β1. By contrast, HCC stroma was negative for both. In addition, besides NCAM, cultured HCC-StCs showed a lower expression of TGF-β1 and of its co-receptor CD105. Interestingly, it has been shown that prostate cancer cells co-cultured with normal StCs showed altered CD105 expression and attenuated TGF-β signalling.50 We do not know if this also occurs in HCC, nor do we know whether the continuous and high production of TGF-β1 by neoplastic hepatocytes in vivo affects (as we have shown here for NCAM) TGF-β-receptor complex on liver StCs inducing a down-modulation of CD105. However, it seems that during HCC progression, liver StCs undergo aberrant changes and lose their typical features and functions which, by contrast, are acquired by neoplastic hepatocytes. In particular, because NCAM has been recently indicated as a distinguishable marker for HCC progenitor cells, that is, liver cancer stem cells,41, 42 the switch of its expression from the liver StCs to neoplastic hepatocyte, may indicate or predict epithelial to mesenchymal transition47 acquiring a more undifferentiated and malignant phenotype. Whether the acquisition of NCAM expression by cirrhotic hepatocytes may prelude to their neoplastic transformation remain to be established. Concomitantly, when losing NCAM, cirrhotic liver StCs acquire a cancer-promoting cell phenotype, because their capacity to support normal vessels morphogeneis (ie, angiogenesis) is also lost. This therefore leads to tumor progression.51 In addition, it remains to be clarified if NCAM expression can be closely associated with a down-modulation of CD105 and if this could be considered another important step to predict epithelial to mesenchymal transition.52, 53 However, our data indirectly support this hypothesis, because nNL-StCs, which are less affected by their proximity to cancer cells than HCC-StCs counterpart, possessed a higher basal level of CD105 and NCAM expression.
Finally, our results show that HCC-StCs are angiogenic powerless, and, most importantly, they release very low amounts of Ang-1, a molecule directly involved in vascular maturation.28, 29 How, and if, these alterations are induced by cancer cells remain to be investigated. However, we cannot exclude that, besides NCAM, even the low level of Ang-1 released by HCC-StCs may help to promote vascular instability in HCC.
In conclusion, tumor stroma has a fundamental role in cancer progression.54 The crosstalk between cancer cells and the tumor stroma is probably mediated through direct cell–cell contact or by several secreted cytokines, growth and profibrotic factors.55 These factors chronically released into the tumor microenvironment may cause significant changes to the resident StCs.55 We here demonstrate that HCC-infiltrating StCs display deeper alterations involving both functional and phenotypic features. Our study, for the first time, underlines the importance of NCAM in inducing abnormal neovascular formations in HCC vascular morphogenesis.56 As vascular normalization in HCC may improve efficacy of chemotherapeutic agents,14 our study provides the rationale for the treatment of HCC patients with chemotherapy and TGF-β1 inhibitors.
References
Folkman J . Role of angiogenesis in tumor growth and metastasis. Semin Oncol 2002;29:15–18.
Nagy JA, Chang SH, Dvorak AM, et al. Why are tumour blood vessels abnormal and why is it important to know? Br J Cancer 2009;100:865–869.
Abramsson A, Berlin O, Papayan H, et al. Analysis of mural cell recruitment to tumor vessels. Circulation 2002;105:112–117.
Morikawa S, Baluk P, Kaidoh T, et al. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 2002;160:985–1000.
von Tell D, Armulik A, Betsholtz C . Pericytes and vascular stability. Exp Cell Res 2006;312:623–629.
Nishishita T, Lin PC . Angiopoietin 1, PDGF-B, and TGF-beta gene regulation in endothelial cell and smooth muscle cell interaction. J Cell Biochem 2004;91:584–593.
Ozerdem U, Stallcup WB . Early contribution of pericytes to angiogenic sprouting and tube formation. Angiogenesis 2003;6:241–249.
Bergers G, Song S . The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol 2005;7:452–464.
Gerhardt H, Betsholtz C . Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res 2003;314:15–23.
Bell RD, Winkler EA, Sagare AP, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010;68:409–427.
Dvorak H . Tumor Blood Vessels in the Endothelium: A Comprehensive Reference. Aird W (ed). Cambridge University Press: Cambridge, 2007.
Jain RK . Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005;307:58–62.
Jain RK . Taming vessels to treat cancer. Sci Am 2008;298:56–63.
Zhu AX, Duda DG, Sahani DV, et al. HCC and angiogenesis: possible targets and future directions. Nat Rev Clin Oncol 2011;8:292–301.
Bruix J, Sherman M, Llovet JM, et al. EASL panel of experts on HCC. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J Hepatol 2001;35:421–430.
Benetti A, Berenzi A, Gambarotti M, et al. Transforming growth factor-beta1 and CD105 promote the migration of hepatocellular carcinoma-derived endothelium. Cancer Res 2008;68:8626–8634.
Seay U, Sedding D, Krick S, et al. Transforming growth factor-beta-dependent growth inhibition in primary vascular smooth muscle cells is p38-dependent. J Pharmacol Exp Ther 2005;315:1005–1012.
Gerhardt H, Wolburg H, Redies C . N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Dev Dyn 2000;218:472–479.
Xian X, Håkansson J, Ståhlberg A, et al. Pericytes limit tumor cell metastasis. J Clin Invest 2006;116:642–651.
Edmonson HA, Steiner PE . Primary carcinoma of the liver. A study of 100 cases among 48.900 necropsies. Cancer 1954;7:462–503.
Gasparini G, Harris AR . Clinical importance of the determination of tumours angiogenesis in breast carcinoma: much more than a new prognostic tool. J Clin Oncol 1995;13:765–782.
Blasi A, Martino C, Balducci L, et al. Dermal fibroblasts display similar phenotypic and differentiation capacity to fat-derived mesenchymal stem cells, but differ in anti-inflammatory and angiogenic potential. Vasc Cell 2011;3:5.
Caruso A, Fiorentini S, Licenziati S, et al. Expansion of rare CD8+ CD28- CD11b- T cells with impaired effectors functions in HIV-1-infected patients. J Acquir Immune Defic Syndr 2000;24:465–474.
Kleinman HK, Martin GR . Matrigel: basament membrane matrix with biological activity. Semin Cancer Biol 2005;15:378–386.
Sawyer JS, Anderson BD, Beight DW, et al. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J Med Chem 2003;46:3953–3956.
Ozerdem U, Grako KA, Dahlin-Huppe K, et al. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn 2001;222:218–227.
Skalli O, Ropraz P, Trzeciak A, et al. A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol 1986;103:2787–2796.
Kim I, Kim HG, Moon SO, et al. Angiopoietin-1 induces endothelial cell sprouting through the activation of focal adhesion kinase and plasmin secretion. Circ Res 2000;86:952–959.
Yu Q, Stamenkovic I . Angiopoietin-2 is implicated in the regulation of tumor angiogenesis. Am J Pathol 2001;158:563–570.
Giuliani N, Colla S, Lazzaretti M, et al. Proangiogenic properties of human myeloma cells: production of angiopoietin-1 and its potential relationship to myeloma-induced angiogenesis. Blood 2003;102:638–645.
Stonāns I, Stonāne E, Russwurm S, et al. HepG2 human hepatoma cells express multiple cytokine genes. Cytokine 1999;11:151–156.
Steingen C, Brening F, Baumgartner L, Schmidt J, et al. Characterization of key mechanism in transmigration and invasion of mesenchymal stem cells. J Mol Cell Cardiol 2008;44:1072–1084.
Tsai JF, Jeng JE, Chuang LY, et al. Clinical evaluation of urinary transforming growth factor b-1 and serum a-fetoprotein as tumor markers of hepatocellular carcinoma. Br J Cancer 1997;75:1460–1466.
Fernández M, Semela D, Bruix J, et al. Angiogenesis in liver disease. J Hepatol 2009;50:604–620.
Ribatti D, Vacca A, Nico B, et al. Angiogenesis and anti-angiogenesis in hepatocellular carcinoma. Cancer Treat Rev 2006;32:437–444.
Fogar P, Basso D, Pasquali C, et al. Neural cell adhesion molecule (N-CAM) in gastrointestinal neoplasias. Anticancer Res 1997;17:1227–1230.
Sasaki H, Yoshida K, Ikeda E, et al. Expression of the neural cell adhesion molecule in astrocytic tumors: an inverse correlation with malignancy. Cancer 1998;82:1921–1931.
Komminoth P, Roth J, Lackie PM, et al. Polysialic acid of the neural cell adhesion molecule distinguishes small cell lung carcinoma from carcinoids. Am J Pathol 1991;139:297–304.
Lantuéjoul S, Laverrière MH, Sturm N, et al. NCAM (neural cell adhesion molecules) expression in malignant mesotheliomas. Hum Pathol 2000;31:415–421.
Volpes R, van den Oord JJ, Desmet VJ . Integrins as differential cell lineage markers of primary liver tumors. Am J Pathol 1993;142:1483–1492.
Xu XL, Xing BC, Han HB, et al. The properties of tumor-initiating cells from a hepatocellular carcinoma patient's primary and recurrent tumor. Carcinogenesis 2010;31:167–174.
Tsuchiya A, Kamimura H, Takamura M, et al. Clinicopathological analysis of CD133 and NCAM human hepatic stem/progenitor cells in damaged livers and hepatocellular carcinomas. Hepatol Res 2009;39:1080–1090.
Geninatti Crich S, Bussolati B, Tei L, et al. Magnetic resonance visualization of tumor angiogenesis by targeting neural cell adhesion molecole with the highly sensitive gadolinium-loaded apoferrin probe. Cancer Res 2006;66:9196–9201.
Bussolati B, Grange C, Bruno S, et al. Neural-cell adhesion molecule (NCAM) expression by immatureand tumor-derived endothelial cells favours cell organization into capillary-like structures. Exp Cell Res 2006;312:913–924.
Gerhardt H, Semb H . Pericytes: gatekeepers in tumour cell metastasis? J Mol Med 2008;86:135–144.
Armilik A, Abramsson A, Betsholtz C . Endothelial/pericyte interaction. Cir Res 2005;97:512–523.
Frame MC, Inman GJ . NCAM is at the heart of reciprocal regulation of E-cadherin and integrin-mediated adhesions via signaling modulation. Dev Cell 2008;15:494–496.
Lehembre F, Yilmaz M, Wicki A, et al. NCAM-induced focal adhesion assembly: a functional switch upon loss of E-cadherin. EMBO J 2008;27:2603–2615.
Cavallaro U . N-cadherin as an invasion promoter: a novel target for antitumor therapy? Curr Opin Investig Drugs 2004;5:1274–1278.
O’Connor JC, Farach-Carson MC, Schneider CJ, et al. Coculture with prostate cancer cells alters endoglin expression and attenuates transforming growth factor-beta signaling in reactive bone marrow stromal cells. Mol Cancer Res 2007;5:585–603.
Bremnes RM, Dønnem T, Al-Saad S, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol 2011;6:209–217.
Fanjul M, Gmyr V, Sengenès C, et al. Evidence for epithelial-mesenchymal transition in adult human pancreatic exocrine cells. J Histochem Cytochem 2010;58:807–823.
Zijlmans HJ, Fleuren GJ, Hazelbag S, et al. Expression of endoglin (CD105) in cervical cancer. Br J Cancer 2009;100:1617–1626.
Bhowmick NA, Moses HL . Tumor-stroma interactions. Curr Opin Genet Dev 2005;15:97–101.
De Wever O, Mareel M . Role of tissue stroma in cancer cell invasion. J Pathol 2003;200:429–447.
Räsänen K, Vaheri A . Activation of fibroblasts in cancer stroma. Exp Cell Res 2010;316:2713–2722.
Acknowledgements
We thank Dr Andrea Smith for editorial assistance. This research was partially supported by AIRC (Associazione Italiana per la Ricerca sul Cancro) Project AIRC IG-9062.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Conventional chemotherapy can be ineffective for hepatocarcinoma (HCC) because of the abnormal structural organization of tumor-infiltrating vessels. The role of TGF-β1 and neural cell adhesion molecule (NCAM) in HCC-induced abnormal vascular morphogenesis are described and provide the rationale for treatment of HCC patients with chemotherapy and TGF-β1 inhibitors.
Supplementary information
Rights and permissions
About this article
Cite this article
Balzarini, P., Benetti, A., Invernici, G. et al. Transforming growth factor-beta1 induces microvascular abnormalities through a down-modulation of neural cell adhesion molecule in human hepatocellular carcinoma. Lab Invest 92, 1297–1309 (2012). https://doi.org/10.1038/labinvest.2012.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2012.94
Keywords
This article is cited by
-
Shed proteoglycans in tumor stroma
Cell and Tissue Research (2016)
-
Variations of circulating endothelial progenitor cells and transforming growth factor-beta-1 (TGF-β1) during thoracic radiotherapy are predictive for radiation pneumonitis
Radiation Oncology (2013)
