
Abstract
MicroRNAs are short noncoding, endogenous RNA species that posttranscriptionally inhibit gene expression by targeting the untranslated region (UTR) of mRNAs. Recently, it was shown that miR-29 inhibits expression of extracellular matrix proteins such as collagens, suggesting an antifibrotic function of miR-29. In the present study, we now investigated the role of miR-29 in profibrogenic growth factor expression as a further central mechanism of fibrosis. Screening of databases revealed putative miR-29 target sequences in the mRNA of platelet-derived growth factor (PDGF)-B, PDGF-B receptor, PDGF-C, vascular endothelial growth factor-A, and insulin-like growth factor (IGF)-I. To analyze miR-29 interaction with the predicted binding sites, we cloned the 3′-UTR sequences of the putative targets in fusion to the luciferase-reporter coding sequence. Functional miR-29 binding to PDGF-C and IGF-I mRNA sequences, but not to the corresponding mutants, was then proven by reporter assays. Hepatic stellate cells (HSC) that transdifferentiate into myofibroblasts, producing extracellular matrix proteins and profibrogenic growth factors, for example, the members of the PDGF family, are crucial for liver fibrosis. Myofibroblastic transition of primary HSC resulted in the loss of miR-29, but in a significant increase of PDGF-C and IGF-I. Compensation of reduced miR-29 levels by miR-29 overexpression in myofibroblastic HSC was followed by a definitive repression of IGF-I and PDGF-C synthesis. After experimental fibrosis, induced by bile-duct occlusion, miR-29 expression was shown to be reduced, but IGF-I and PDGF-C expression was upregulated, correlating inversely to the miR-29 pattern. Thus, we conclude that miR-29, downregulated during fibrosis, acts as an antifibrogenic mediator not only by targeting collagen biosynthesis, but also by interfering with profibrogenic cell communication via PDGF-C and IGF-I.
Similar content being viewed by others


Main
MicroRNAs (miRNAs) are small noncoding RNA molecules that posttranscriptionally regulate gene expression. In human, more than thousand miRNA species are known.1 Interaction of miRNAs with the untranslated region (UTR), most often with the 3′-UTR, of various transcripts results in transcript degradation or translational repression.2 In a wide range of cancer types, miRNAs are dysregulated, affecting cell proliferation and differentiation because of their impact on gene regulation.3 In hepatocellular carcinoma (HCC), alteration of miRNA expression profiles are suggested to be involved in increased proliferation, liver-tumor initiation, and progression.4, 5, 6 Furthermore, divergent miRNA patterns were observed during chronic liver disease due to various etiologies,7, 8, 9 but function of miRNA in chronic inflammatory liver diseases is not well known.10, 11 Chronic liver disease, independent of the causing noxa, results in fibrosis, which is characterized by excessive accumulation of extracellular matrix (ECM) proteins, in particular, of various collagens.12, 13 The central cell type of liver fibrosis are myofibroblastic cells of different origin, producing mainly the ECM components and being responsible for profibrogenic growth factor secretion.12, 14, 15 Sinusoidal hepatic stellate cells (HSC) are the main source of myofibroblastic cells and matrix deposition.12, 16 In the quiescent state, they store 80% of the body vitamin A in fat droplets. After liver injury, however, they lose fat droplets, get activated, and transdifferentiate into myofibroblasts. Myofibroblastic transition is characterized by an increased α-smooth muscle actin (SMA) assembly, augmented matrix production, and by an enhanced profibrotic growth factor secretion.12, 16, 17 This comprehensive change in gene expression is associated with a pronounced dysregulation of miRNA synthesis.18, 19, 20 Recent studies have shown that several miRNA, such as miR-16,21 miR-27a/b,20 miR-195,22 and miR-33523 are involved in proliferation or induced migration of myofibroblastic-activated HSC. Others such as miR-19b, miR-150, and miR-194 are suggested to participate in TGF-β signaling and myofibroblastic HSC activation, respectively.24, 25 In particular, members of the miR-29 family, repressed during myofibroblastic activation,26 are of special interest, because their dysregulation has been shown to be involved in synthesis of ECM proteins.26, 27, 28, 29, 30
The miR-29 family consists of miR-29a, miR-29b, and miR-29c, differing only in two or three bases. The miR-29a and miR-29b1, as well as miR-29c and miR-29b2, are encoded and transcribed in tandem by two genes located on chromosome 7 or chromosome 1, respectively.31, 32 van Rooij et al30 first emphasized the role of miR-29 after myocardial infarction, demonstrating its inhibitory effect on elastin, collagen I and III synthesis in cardiac fibroblasts, and its repression by TGF-β. Accordingly, downregulation of miR-29 was suggested to enhance the fibrotic response after myocardial infarction, whereas overexpression of miR-29 in cardiac fibroblasts reduced collagen expression. Similarly, Ogawa et al29 demonstrated that miR-29 inhibits the production of fibrillar collagen in HSC, suggesting also a function of miR-29 in liver fibrosis.26
In the present study, we provide detailed evidence for the antifibrotic action of miR-29 interfering with the profibrotic growth factor release from HSC, pointing to its central role in chronic liver disease. Our data demonstrate that miR-29 represses not only fibrotic accumulation of collagen I, III, and IV as previously shown,26, 27, 28, 29, 30 but also expression of profibrogenic mediators, platelet-derived growth factor C (PDGF-C) and insulin-like growth factor I (IGF-I) in HSC.
MATERIALS AND METHODS
Bioinformatics
To identify targets of miR-29 the databases Miranda (http://www.microrna.org),33 Targetscan (http://www.targetscan.org),34 and Pictar (http://pictar.bio.nyu.edu)35 were screened. Common target sequences of miR-29, identified by the algorithms of Miranda, Pictar, and Targetscan, were evaluated using the query platform of the Mathematical Bioscience Institute at Ohio State University, Columbus, OH, USA; (http://www.mbi.osu.edu) and listed according to their ranking score.
Plasmid Construction
Targets of miR-29 were queried using different databases as described in Table 1. The putative binding sites of PDGF-C, vascular endothelial growth factor (VEGF)-A, and IGF-I, as well as the corresponding mutants containing two point mutations were created by oligonucleotide dimerization and subsequent insertion downstream to the Renilla luciferase gene of the psiCHECK-2 vector (Ambion, Austin, TX, USA; Figure 2a). Oligonucleotides used for dimerization are listed in Supplementary Table S1.
Isolation, Primary Cell Culture, and Stimulation of HSC
Isolation of primary rat HSC was performed by liver perfusion and Nycodenz density centrifugation as previously described.17, 36 For myofibroblastic transition, HSC were maintained in Dulbecco's modified Eagles medium (DMEM) with 20% fetal calf serum (FCS) the first 2 days after isolation, followed by culture in DMEM with 10 % FCS. Myofibroblastic transition was characterized by induction of α-SMA expression. HSC at day 3 are considered as quiescent relative to day 7 of HSC culture, expressing high levels of SMA, and considered as myofibroblastic cells.
HSC-T6, kindly provided by SL Friedman, were cultured as previously described,37 and used for miR-29 transfection. As a positive control for miR-29-induced translational inhibition, cells were treated with 1 μg/ml cycloheximide. After 24 h, cells were lysed for subsequent protein or RNA analyses, respectively. All cells were maintained at 37 °C and 5% CO2 in a humidified atmosphere.
Total RNA Extraction
Total RNA from snap-frozen cell culture, tissue, and serum samples was isolated using the Qiazol reagent following the instructions of the supplier (Qiagen, Hilden, Germany). RNA quantity was determined by A260-measurement using the ND-1000 NanoDrop spectrophotometer (NanoDrop, Wilmington, DE, USA), quality was assessed by microcapillary electrophoresis (2100 BioAnalyser, Agilent Technologies, Waldbronn, Germany), and RNA preparations from human formalin-fixed and paraffin-embedded biopsies were performed as described previously.38
Quantifying miRNA by Real-Time PCR
miRNA was analyzed by a two-step real-time PCR using the miScript-Reverse Transcription Kit and the miRNA-SYBR Green PCR Kit (Qiagen). Primers used for cDNA synthesis and real-time PCR were selected and purchased from the GeneGlobe Search Center (Qiagen). All steps were performed in triplicate and in agreement with the supplier's guidelines. Cellular miRNA levels were normalized using RNU6 as reference.
Transcript Quantification by Real-Time PCR
Total RNA (1 μg) was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems Applera, Darmstadt, Germany) as instructed. A total of 10 ng cDNA were used for real-time PCR using the Power SYBR Green PCR Mastermix (Applied Biosystems Applera), and specific primers listed in the Supplementary Table S2. Transcript levels were evaluated by absolute quantification using an online standard curve and corrected by normalization to the house-keeping gene hypoxanthin-phosphoribosyl-transferase (HPRT).
miRNA Transfection and Reporter Assays
miRNA mimicking miR-29a, miR-29b, or a scrambled miRNA control were obtained from Dharmacon (Lafayette, CO, USA) and used for transfection of HSC-T6 cells by lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) following the instructions of the supplier. Luciferase levels were measured in triplicate by means of the Dual Luciferase Reporter Assay System (Promega, Mannheim, Germany) according to the manufacturer's recommendations.
Immunoblotting and ELISA
Equal amounts of protein (10 μg) were resolved on 4–12% SDS-polyacrylamide gels (Biorad, München, Germany) and used for immunoblotting. Anti-PDGF-C (1:300) was purchased from Santa Cruz (Heidelberg, Germany). For quantification of rat IGF-I in the HSC medium, an IGF-I immunoassay (R&D Systems, Wiesbaden-Nordenstadt, Germany) was used. Rat VEGF-A was measured in cell culture supernatants with a rat VEGF ELISA kit (RayBiotech, Norcross, GA, USA) as instructed.
Induction of Biliary Fibrosis after Bile-Duct Occlusion in Rats
A total of 12 Wistar rats (250–300 g) were subjected to bile-duct occlusion (BDO) as described earlier,39, 40 whereas 8 animals were sham-operated. All experiments were conducted in accordance with the National Health and Medical Research Committee Guidelines for Animal Experimentation. Livers were snap-frozen in liquid nitrogen, and used for RNA analysis and hydroxyproline determination as described before.40 Liver tissue from liver segment IV was fixed in 4% buffered paraformaldehyde and embedded in paraffin for histopathological evaluation after Gomori and Sirius Red staining.
Statistical Analysis
Statistical analyses were performed using SPSS software 17 (Chicago, IL, USA). Data obtained from rat tissues with normal distribution (Kolmogorov–Smirnov test) was subjected to analyses of variance using Dunnett's post-hoc comparison, to test for differences in means of variables. Bar graphs show means±s.e.m. For correlation of miR-29 expression with fibrotic features, the Spearman's rho-coefficient was calculated and verified by the two-tailed significance test.
RESULTS
The PDGF-C and IGF-I Transcripts are Novel Targets of miR-29
Previous reports have demonstrated that miR-29 targets expression of matrix components, especially the expression of collagen subunits.26, 29, 30 Our query of databases revealed that mRNA of members of the PDGF ligand and receptor family, and IGF-I might be further putative targets of miR-29. The miR-29 binding sites were predicted in the 3′-UTR of the PDGF-BB receptor, PDGF-C and VEGF-A, and in the IGF-I mRNA (Table 1). All of these putative binding sites were highly conserved between species (Figure 1), making it even more likely that they might represent regulatory targets of miR-29.

Conservation of putative binding sites in the 3′-untranslated region (UTR) of platelet-derived growth factor (PDGF)-B, PDGF-C, insulin-like growth factor (IGF)-I, and vascular endothelial growth factor (VEGF)-A mRNA. Sequence alignment of putative miR-29 binding sites of PDGF-B, PDGF-C, IGF-I, and VEGF-A show significant conservation of the interaction sites between species such as Mus musculus (Mmu), Homo sapiens (Hsa), Armadillo, Dasypus novemcinctus (Dno), Gallus gallus (Gga), Rattus norvegicus (Rno), Tenrec, Echinops telfairi (Ete), or Xenopus tropicalis (Xtr). The interaction sites of the miR-29 seed sequence are indicated in white. Numbers in the target sequences show the position of the potential miR-29 binding sites in the 3′-UTR of the respective mRNA.
Specific miR-29 Interaction to the 3′-UTR of PDGF-C and IGF-A Transcripts
To analyze miR-29 binding to the UTR of these putative targets, we inserted either the predicted interaction site or the corresponding sequence, carrying two point mutations, downstream to the luciferase reporter (Figure 2a). Indeed, luciferase reporter assays confirmed the interaction of miR-29 with the 3′-UTR of PDGF-C, VEGF-A, and IGF-I (Figures 2c–e), but not with the 3′-UTR of PDGF-BB receptor or PDGF-BB (data not shown). The specificity of miR-29 interaction with the 3′-UTR binding sites of PDGF-C, IGF-I, and VEGF-A was proven by the marked repression of the wild-type reporter constructs in miR-29-treated HSC, whereas no miR-29 inhibitory effects were observed on the activities of reporters carrying mutated binding sites (Figures 2c–e).

Interaction of miR-29 with the 3′-untranslated region (UTR) of platelet-derived growth factor (PDGF)-C, insulin-like growth factor (IGF)-I, and vascular endothelial growth factor (VEGF)-A transcripts. Putative binding sites of PDGF-C, IGF-I, and VEGF-A were subcloned downstream of the luciferase reporter (hRluc) of the psiCHECK-2 vector (a) and transfected into hepatic stellate cells (HSC)-T6 cells. As a positive control, a miR-29 complementary sequence (miR-29 consensus) inserted downstream of the hRluc reporter was used as miR-29 sensor (b). Reporter plasmids, including putative miR-29 target sequences of the 3′-UTR of PDGF-C (c), IGF-I (d), and VEGF-A (e) transcripts, and the corresponding mutated sequences carrying two point mutations (bold and underlined), were co-transfected into HSC-T6 in combination with scrambled microRNA (miRNA) or miR-29 mimics (ago-miR-29a), respectively (c–e). Luciferase reporter expression was determined by the hRluc luminescence measurement normalized to firefly luminescence (hluc+). Interaction of miR-29 with functional binding sites in the reporter constructs decreased luciferase reporter gene expression (b–e) (*P<0.05, **P<0.01).
Synthesis of PDGF-C and IGF-I in HSC Is Inhibited by Overexpression of miR-29
Both members of the miR-29 family, miR-29a and miR-29b, are decreased during myofibroblastic transition of primary HSC (Figure 3a), whereas the expression levels of putative targets PDGF-C and IGF-I are markedly increased. In contrast to PDGF-C and IGF-I, the expression of VEGF-A, also identified as a putative target by bioinformatics and belonging to the PDGF-family, is not upregulated during myofibroblastic transition (Figure 3b).

miR-29 decrease, but platelet-derived growth factor (PDGF)-C and insulin-like growth factor (IGF)-I increase during myofibroblastic transition of hepatic stellate cells (HSC). miR-29a and miR-29b (a), and the potential miR-29 targets PDGF-C, IGF-I, and vascular endothelial growth factor (VEGF)-A (b) were quantified in total RNA extracted from rat HSC at day 3, 5, 7, and 30 (3d, 5d, 7d, and 30d) of primary culture (*P<0.05, **P<0.01).
To compensate for the reduced levels of miR-29 members in myofibroblastic HSC, we treated HSC-T6 cells, representing the myofibroblastic HSC phenotype, with HGF. Our recent findings have shown that HGF upregulates miR-29a and miR-29b up to 3- to 4-fold in both primary HSC and myofibroblastic HSC-T6 cells.27 The HGF treatment of HSC-T6 cells resulted in a definitive decrease of PDGF-C and IGF-I expression (Figure 4). Next, we overexpressed miR-29a and miR-29b by transfection of the respective mimics into HSC-T6 cells, leading to a 100-fold increase of each miR-29 member in HSC (Supplementary Figure S1). This miR-29 overexpression caused a marked repression of PDGF-C and IGF-I transcripts in myofibroblastic HSC, but not of VEGF-A mRNA levels (Figure 5a).

Reduced expression of platelet-derived growth factor (PDGF)-C and insulin-like growth factor (IGF)-I in hepatic stellate cells (HSC) after HGF treatment. HSC-T6 cells were treated with HGF, and PDGF-C, IGF-I, and VEGF-A mRNA were quantified by real-time PCR. PDGF-C, IGF-I, and VEGF-A mRNA levels were normalized using hypoxanthin-phosphoribosyl-transferase (HPRT) and indicated as relative units (RE) (*P<0.05, **P<0.01).

Influence of miR-29a and miR-29b on platelet-derived growth factor (PDGF)-C, insulin-like growth factor (IGF)-I, and vascular endothelial growth factor (VEGF)-A synthesis. Myofibroblastic hepatic stellate cell (HSC)-T6 cells treated with either miR-29a, miR-29b (ago-miR-29a, ago-miR-29b), scrambled microRNA (miRNA), or cycloheximide were analyzed for transcript (a) and protein (b) levels of PDGF-C, IGF-I, and VEGF-A. All transcript levels were quantified by real-time PCR (a). PDGF-C protein synthesis was evaluated by three independent western immunoblotting experiments and subsequent densitometric quantification (relative integrated densitometric units, IDV). Protein values of IGF-I and VEGF-A were quantified by ELISA (b) (*P<0.05, **P<0.01).
Inhibition of IGF-I and PDGF-C protein synthesis by miR-29 was then compared with translational inhibition by cycloheximide, emphasizing the definitive inhibitory influence of miR-29 on IGF-I and PDGF-C synthesis, whereas VEGF-A synthesis was not affected (Figure 5b). Thus, our data show for the first time that miR-29 is not only involved in preventing fibrosis through the repression of ECM synthesis, but also by repressing PDGF-C and IGF-I.
High IGF-I and PDGF-C Expression in Liver Tissues Correlates with Reduced miR-29 Levels after Experimental Fibrosis
The miR-29 expression was previously shown to be reduced in liver specimens undergoing experimental fibrosis.26, 27 To analyze the link between PDGF-C and IGF-I expression, and reduced miR-29 levels in the fibrotic liver, we performed BDO in rats, and miR-29 levels were compared with miR-29 levels in livers of sham-operated animals. Four weeks after BDO, fibrotic septa had developed (Figure 6a), accompanied by a marked decrease in miR-29a and miR-29b levels (Supplementary Figure S2) as shown recently.26, 27 Furthermore, evidence for inflammation and elevated collagen synthesis was found in the fibrotic livers as determined by recruitment of inflammatory cells, Gomori histology, and by rising hydroxyproline levels. Both, the inflammatory response and the increase of matrix production significantly correlated with a decrease in the levels of miR-29 (Figure 6b).

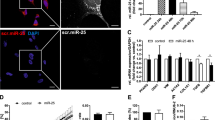
Platelet-derived growth factor (PDGF)-C and insulin-like growth factor (IGF)-I in experimental liver fibrosis after bile-duct occlusion (BDO) in rats. Hepatic fibrosis was detected by Gomori staining and inflammation by chloroacetate esterase (CA) granulocyte staining (red) in sham-operated (sham) (N=8) and BDO rat livers (N=12) (a). Inverse miR-29a correlation with inflammation and hydroxyproline levels after BDO-induced fibrosis (b). PDGF-C and IGF-I increase after experimental fibrosis; PDGF-C and IGF-I expression was determined in sham-operated and BDO treated rats by real-time PCR normalized to hypoxanthin-phosphoribosyl-transferase (HPRT) values (c) (*P<0.05, **P<0.01).
As it has been suggested by previous reports that there might be a systematic inverse relationship between the levels of miRNA expression and the potential target transcripts,41, 42 we analyzed if the reduced miR-29 levels are associated with increased PDGF-C, IGF-I, and VEGF-A mRNA levels (Figure 6c). Indeed, examination of PDGF-C, IGF-I, and VEGF-A mRNA levels in comparison with the miR-29 expression demonstrates that the reduction of miR-29 after experimental fibrosis in rats is associated with enhanced expression of PDGF-C and IGF-I (Figure 6c, Table 1), though the inverse correlation for PDGF-C was rather weak. In agreement with the in-vitro studies in HSC, VEGF-A expression was not inversely correlated with miR-29 levels in hepatic fibrosis (Table 2).
DISCUSSION
In this study, we provide evidence for a prominent role of miR-29 as a central antifibrotic mediator of liver fibrogenesis, affecting not only ECM production, but also expression of the profibrogenic growth factors PDGF-C and IGF-I.
On the basis of our in-silico data, miR-29 is predicted to interact with a number of mRNAs encoding for cellular signal transducers and growth factors. Accordingly, previous reports have emphasized its role in carcinogenesis, demonstrating post-transcriptional repression of mRNA involved in cell cycle control, apoptosis,31, 43, 44 and DNA methylation.45 In terms of fibrosis, however, miR-29 has so far only been described as a prominent regulator of the ECM synthesis.26, 27, 28, 29, 30
As myofibroblastic HSC express a wide panel of cytokines and growth factors that perpetuate the fibrotic process through paracrine and autocrine effects,12, 46 we specifically investigated profibrotic mediators as novel putative targets of miR-29. Previous studies of Sekiya et al47 indicate that miR-29b represses myofibroblastic activation, including synthesis of SMA and PDGF-BB receptor. Although our in-silico analysis suggested that miR-29 might regulate a large mRNA cluster of the PDGF ligand and receptor family, our functional analyses provided little evidence for posttranscriptional inhibition of the PDGF-BB receptor, PDGF-B, and VEGF-A, by miR-29. Instead, our miR-29 binding studies and expression data of miR-29-treated HSC clearly demonstrated that the growth factors PDGF-C and IGF-I were tightly regulated by miR-29. These novel targets of miR-29 are known to have prominent roles in liver fibrogenesis by triggering HSC proliferation and their transition into myofibroblasts, which is accompanied by extensive collagen synthesis.48, 49 However, miR-29 treatment of other cell types, such as embryonic muscle cells also results in a reduction of IGF-I and PDGF-C (data not shown), suggesting a general function of miR-29 in synthesis of these growth factors. Therefore, miR-29 impedes the ECM accumulation during liver fibrogenesis not only by direct suppression of collagen synthesis, but also by interfering with the PDGF-C- and IGF-I-mediated profibrotic pathways. Although hepatocytes are the main source of IGF-I, leading to paracrine stimulation of HSC during fibrogenesis, autocrine stimulation is also induced by IGF-I upregulation during myofibroblastic transdifferentiation. Although IGF-I has also been shown to induce apoptosis in HSC,50 in cooperation with PDGF-BB, it certainly functions as an important mitogen for hepatic myofibroblasts.51, 52 Accordingly, miR-29, shown to affect moderately HSC proliferation,53 might be involved in HSC growth by posttranscriptional IGF-I synthesis control.
The other functional target of miR-29, PDGF-C, belongs to the PDGF growth factor family and binds to the PDGF receptor-A dimers or to PDGF receptor-AB heterodimers.54, 55 PDGF-C elicits mitogenic and migratory activities in fibroblasts, smooth muscle cells, and pericytes,54, 55 and is highly upregulated in myofibroblastic HSC.48 Its hepatic overexpression in a transgenic mouse model produced histomorphological changes ranging from steatosis to steatohepatitis and fibrosis, ultimately leading to hepatocellular carcinogenesis.56 IGF-I is also known to have an important role in hepatocarcinogenesis.57 As miR-29 is reduced in HCC,44 overexpression of PDGF-C and IGF-I during hepatocarcinogenesis56, 57 might be a consequence of miR-29 dysregulation.
The antifibrotic function of miR-29 in experimental and human fibrosis is further underlined by its inverse correlation with the severity of inflammation and fibrosis.27 van Rooij et al30 have previously studied the function of miR-29 in myocarditis, pointing out that TGF-β is a main suppressor of miR-29 expression. We showed recently that both central profibrotic factors, PDGF-BB and TGF-β, repress miR-29 in HSC, and might be responsible for the reduction of miR-29 in the fibrotic liver.27 Therefore, miR-29 is a crucial mediator in cellular communication pathways of liver fibrosis, being regulated by profibrogenic growth factors on one hand and controlling the synthesis of PDGF-C and IGF-I on the other.
References
Kozomara A, Griffiths-Jones S . miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 2011;39, (database issue) D152–D157.
Bartel DP . MicroRNAs: target recognition and regulatory functions. Cell 2009;2:215–233.
Ambros V . The functions of animal microRNAs. Nature 2004;7006:350–355.
Varnholt H, Drebber U, Schulze F, et al. MicroRNA gene expression profile of hepatitis C virus-associated hepatocellular carcinoma. Hepatology 2008;4:1223–1232.
Gramantieri L, Fornari F, Callegari E, et al. MicroRNA involvement in hepatocellular carcinoma. J Cell Mol Med 2008;6A:2189–2204.
Braconi C, Henry JC, Kogure T, et al. The role of microRNAs in human liver cancers. Semin Oncol 2011;6:752–763.
Cheung O, Puri P, Eicken C, et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008;6:1810–1820.
Jin X, Ye YF, Chen SH, et al. MicroRNA expression pattern in different stages of nonalcoholic fatty liver disease. Dig Liver Dis 2009;4:289–297.
Morita K, Taketomi A, Shirabe K, et al. Clinical significance and potential of hepatic microRNA-122 expression in hepatitis C. Liver Int 2011;4:474–484.
Chang Y, Jiang HJ, Sun XM, et al. Hepatic stellate cell-specific gene silencing induced by an artificial microRNA for antifibrosis in vitro. Dig Dis Sci 2010;3:642–653.
Lakner AM, Bonkovsky HL, Schrum LW . microRNAs: fad or future of liver disease. World J Gastroenterol 2011;20:2536–2542.
Friedman SL . Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;6:1655–1669.
Schuppan D, Krebs A, Bauer M, et al. Hepatitis C and liver fibrosis. Cell Death Differ 2003;10:S59–S67.
Kisseleva T, Uchinami H, Feirt N, et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol 2006;3:429–438.
Knittel T, Kobold D, Saile B, et al. Rat liver myofibroblasts and hepatic stellate cells: different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology 1999;5:1205–1221.
Schuppan D, Porov Y . Hepatic fibrosis: from bench to bedside. J Gastroenterol Hepatol 2002;17:S300–S305.
Ramadori G, Veit T, Schwogler S, et al. Expression of the gene of the alpha-smooth muscle-actin isoform in rat liver and in rat fat-storing (ITO) cells. Virchows Arch B Cell Pathol Incl Mol Pathol 1990;6:349–357.
Mann J, Chu DC, Maxwell A, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010;2:705–714 714 e1–4.
Guo CJ, Pan Q, Cheng T, et al. Changes in microRNAs associated with hepatic stellate cell activation status identify signaling pathways. FEBS J 2009;18:5163–5176.
Ji J, Zhang J, Huang G, et al. Over-expressed microRNA-27a and 27b influence fat accumulation and cell proliferation during rat hepatic stellate cell activation. FEBS Lett 2009;4:759–766.
Guo CJ, Pan Q, Jiang B, et al. Effects of upregulated expression of microRNA-16 on biological properties of culture-activated hepatic stellate cells. Apoptosis 2009;11:1331–1340.
Sekiya Y, Ogawa T, Iizuka M, et al. Down-regulation of cyclin E1 expression by microRNA-195 accounts for interferon-beta-induced inhibition of hepatic stellate cell proliferation. J Cell Physiol 2010;10:2535–2542.
Chen C, Wu CQ, Zhang ZQ, et al. Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation. Exp Cell Res 2011;12:1714–1725.
Lakner AM, Steuerwald NM, Walling TL, et al. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology 2012 (in press).
Venugopal SK, Jiang J, Kim TH, et al. Liver fibrosis causes down-regulation of miRNA-150 and miRNA-194 in hepatic stellate cells and their over-expression causes decreased stellate cell activation. Am J Physiol Gastrointest Liver Physiol 2010;1:G101–G106.
Roderburg C, Urban GW, Bettermann K, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 2011;1:209–218.
Kwiecinski M, Noetel A, Elfimova N, et al. Hepatocyte growth factor (HGF) inhibits collagen I and IV synthesis in hepatic stellate cells by miRNA-29 induction. PLoS One 2011;9:e24568.
Maurer B, Stanczyk J, Jungel A, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum 2010;6:1733–1743.
Ogawa T, Iizuka M, Sekiya Y, et al. Suppression of type I collagen production by microRNA-29b in cultured human stellate cells. Biochem Biophys Res Commun 2010;1:316–321.
van Rooij E, Sutherland LB, Thatcher JE, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA 2008;35:13027–13032.
Mott JL, Kobayashi S, Bronk SF, et al. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007;42:6133–6140.
Wang H, Garzon R, Sun H, et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008;5:369–381.
John B, Enright AJ, Aravin A, et al. Human MicroRNA targets. PLoS Biol 2004;11:e363.
Creighton CJ, Nagaraja AK, Hanash SM, et al. A bioinformatics tool for linking gene expression profiling results with public databases of microRNA target predictions. Rna 2008;11:2290–2296.
Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet 2005;5:495–500.
Geerts A, Vrijsen R, Rauterberg J, et al. In vitro differentiation of fat-storing cells parallels marked increase of collagen synthesis and secretion. J Hepatol 1989;1:59–68.
Vogel S, Piantedosi R, Frank J, et al. An immortalized rat liver stellate cell line (HSC-T6): a new cell model for the study of retinoid metabolism in vitro. J Lipid Res 2000;6:882–893.
Siebolts U, Varnholt H, Drebber U, et al. Tissues from routine pathology archives are suitable for microRNA analyses by quantitative PCR. J Clin Pathol 2009;1:84–88.
Boigk G, Stroedter L, Herbst H, et al. Silymarin retards collagen accumulation in early and advanced biliary fibrosis secondary to complete bile duct obliteration in rats. Hepatology 1997;3:643–649.
Tox U, Scheller I, Kociok N, et al. Expression of angiotensin II receptor type 1 is reduced in advanced rat liver fibrosis. Dig Dis Sci 2007;8:1995–2005.
Lim LP, Lau NC, Garrett-Engele P, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005;7027:769–773.
Wang X, Wang X . Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res 2006;5:1646–1652.
Park SY, Lee JH, Ha M, et al. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol 2009;1:23–29.
Xiong Y, Fang JH, Yun JP, et al. Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology 2010;3:836–845.
Fabbri M, Garzon R, Cimmino A, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA 2007;40:15805–15810.
Reeves HL, Friedman SL . Activation of hepatic stellate cells--a key issue in liver fibrosis. Front Biosci 2002;7:d808–d826.
Sekiya Y, Ogawa T, Yoshizato K, et al. Suppression of hepatic stellate cell activation by microRNA-29b. Biochem Biophys Res Commun 2011;1:74–79.
Breitkopf K, Roeyen C, Sawitza I, et al. Expression patterns of PDGF-A, -B, -C and -D and the PDGF-receptors alpha and beta in activated rat hepatic stellate cells (HSC). Cytokine 2005;5:349–357.
Svegliati-Baroni G, Ridolfi F, Di Sario A, et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways. Hepatology 1999;6:1743–1751.
Saile B, DiRocco P, Dudas J, et al. IGF-I induces DNA synthesis and apoptosis in rat liver hepatic stellate cells (HSC) but DNA synthesis and proliferation in rat liver myofibroblasts (rMF). Lab Invest 2004;8:1037–1049.
Bridle KR, Li L, O′Neill R, et al. Coordinate activation of intracellular signaling pathways by insulin-like growth factor-1 and platelet-derived growth factor in rat hepatic stellate cells. J Lab Clin Med 2006;5:234–241.
Novosyadlyy R, Dudas J, Pannem R, et al. Crosstalk between PDGF and IGF-I receptors in rat liver myofibroblasts: implication for liver fibrogenesis. Lab Invest 2006;7:710–723.
Bandyopadhyay S, Friedman RC, Marquez RT, et al. Hepatitis C virus infection and hepatic stellate cell activation downregulate miR-29: miR-29 overexpression reduces hepatitis C viral abundance in culture. J Infect Dis 2011;12:1753–1762.
Gilbertson DG, Duff ME, West JW, et al. Platelet-derived growth factor C (PDGF-C), a novel growth factor that binds to PDGF alpha and beta receptor. J Biol Chem 2001;29:27406–27414.
Li X, Ponten A, Aase K, et al. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat Cell Biol 2000;5:302–309.
Campbell JS, Hughes SD, Gilbertson DG, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci USA 2005;9:3389–3394.
Scharf JG, Dombrowski F, Ramadori G . The IGF axis and hepatocarcinogenesis. Mol Pathol 2001;3:138–144.
Acknowledgements
This study was supported by the Research and Education program of the Medical Faculty of the University of Cologne and by the German Competence Network for Viral Hepatitis (HepNet), funded by the German Ministry of Education and Research (BMBF), Grant No. 01KI0601 (to HPD/MO) and the German Liver Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
A novel mechanism for the antifibrogenic effects of miR-29 in the liver is presented. Expression of miR-29 is decreased in hepatic stellate cells (HSC) transdifferentiating into myofibroblasts during liver fibrogenesis. Overexpression of miR-29 inhibits profibrogenic IGF-I and PDGF-C expression in myofibroblastic HSC through posttranscriptional mRNA interaction, in addition to its known effects on collagen biosynthesis.
Rights and permissions
About this article
Cite this article
Kwiecinski, M., Elfimova, N., Noetel, A. et al. Expression of platelet-derived growth factor-C and insulin-like growth factor I in hepatic stellate cells is inhibited by miR-29. Lab Invest 92, 978–987 (2012). https://doi.org/10.1038/labinvest.2012.70
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2012.70
Keywords
This article is cited by
-
The roles of microRNA families in hepatic fibrosis
Cell & Bioscience (2017)
-
MicroRNA-29: A Crucial Player in Fibrotic Disease
Molecular Diagnosis & Therapy (2017)
-
MicroRNAs in fibrosis: opportunities and challenges
Arthritis Research & Therapy (2016)
-
MiR-29a Assists in Preventing the Activation of Human Stellate Cells and Promotes Recovery From Liver Fibrosis in Mice
Molecular Therapy (2016)
-
RETRACTED ARTICLE: Tissue expression levels of miR-29b and miR-422a in children, adolescents, and young adults’ age groups and their association with prediction of poor prognosis in human osteosarcoma
Tumor Biology (2016)

{kind=link}
{kind=link}