
Abstract
Polo-like kinase 1 (Plk1) is a mitotic serine/threonine kinase and its kinase activity is closely interrelated to cell cycle progression, various types of cancer development and often correlates with poor prognosis. Thus, it is of prime importance to characterize the phenotypes of Plk1 inhibition in cells for drug development and clinical application. Here, we report a novel kinase inhibitory chemical, CBB2001, which specifically inhibited Plk1 kinase activity in vitro with an IC50 of 0.39 μM. In cervical carcinoma HeLa cells, we found that treatment of CBB2001 caused mitotic cell cycle arrest (EC50=0.72 μM) and induction of ‘polo’ cells (EC50=0.32 μM). Interestingly, the cell cycle arrest induced by CBB2001 was associated with accumulation of Plk1 (EC50=0.61 μM) and Geminin (EC50=0.43 μM) proteins, but distinct from the phenotypes induced by Aurora kinase inhibitors. The inhibitory effects of CBB2001 were phenocopied by RNA interferences of Plk1. We also confirmed the cell cycle inhibitory effects of CBB2001 in other cancer cells. Moreover, CBB2001 inhibited the growth of HeLa cells with an IC50 of 0.85 μM in MTT assays, which is better than that of reported Plk1 inhibitory chemicals ON01910 (IC50=6.46 μM) and LFM-A13 (IC50=37.36 μM). CBB2001 also inhibited mouse xenograft tumor growth. Furthermore, CBB2001 inhibited mitotic exit and delayed degradation of APC/C substrates, Geminin, Cyclin B1 and Aurora A. These specific phenotypes may serve as specific features for Plk1 inhibition and for Plk1-based clinic trials.
Similar content being viewed by others


Main
Polo-like kinase 1 (Plk1) is a mitosis-specific serine/threonine kinase that has a critical role in mitosis.1, 2, 3 It has been shown that Plk1 is required for the activation of cyclin B/CDK1 during the progression of G2 phase to M-phase, cohesin removal from the arms of chromosomes for sister chromatid separation in mitosis, activation of the APC/C ubiquitin E3 ligase complex, assembly of mitotic spindles, mitotic exit and cytokinesis.1, 2, 3 Plk1 has been reported to phosphorylate many important cell cycle regulators, such as the components of the cohesin complex, Cdc25C, Myt1 and Emi1.1, 2, 3, 4, 5, 6, 7 Loss of Plk1 prevents chromosome segregation during mitosis and induces mitotic arrest.8, 9, 10, 11 Recently, Plk1 has been shown to phosphorylate HBO1, a histone acetyltransferase that regulates the loading of the critical replication licensing protein, CDT1, onto chromatin in mitosis and alteration of HBO1 phosphorylation affects the next round of DNA replication.12 Plk1 expression is associated with proliferation of cells and it is overexpressed in a variety of cancers, including breast, bladder, prostate, pancreatic, papillary thyroid, ovarian and head and neck cancers, as well as non-small-cell lung carcinomas and non-Hodgkin’s lymphomas.13, 14, 15, 16 The overexpression of Plk1 is often associated with poor prognosis and low overall survival. Downregulation of Plk1 activity, either by siRNA- or Plk1-specific chemical inhibitors, has been shown to prevent cell proliferation in cancer cell lines or in tumor xenografts.17, 18 Plk1 thus represents a promising therapeutic target for cancer intervention.
Plk1 is activated by phosphorylation at its threonine 210 (T210), but the kinase(s) that phosphorylates Plk1 T210 remains elusive. Recently, it was found that Aurora A kinase, another critical mitotic serine/threonine kinase, together with Bora, phosphorylates T210 in Plk1 to activate Plk1 during S phase to G2/M transition.19, 20 Aurora kinases (Aurora A and Aurora B) are key mitotic regulators that are involved in centrosome duplication, formation of bipolar mitotic spindle, chromosome alignment in mitotic spindles and spindle checkpoint control.21, 22 The gene encoding Aurora A kinase is frequently amplified in human colorectal cancers and other cancers, and the Aurora kinase proteins are also overexpressed in many human cancers.23, 24 Expression of Aurora A kinase in normal cells is sufficient to induce genome instability and oncogenic transformation.23, 24 The involvement of Aurora kinases in activating Plk1 suggests that the inhibitors of Aurora kinases may be used for human cancers that overexpress Plk1. To evaluate this possibility, we have developed a new Plk1 inhibitory chemical and analyzed the effects of Plk1 and Aurora kinase inhibitors.
The newly developed kinase inhibitory chemical CBB2001 efficiently inhibits Plk1 kinase activity both in vitro and in vivo, with an IC50 of 0.85 μM for tumor cell growth inhibition. CBB2001 also induces mitotic ‘polo’ phenotype in various types of cancer cells, which is associated with Plk1 protein accumulation and appearance of the modified band of Geminin, an endogenous inhibitor of DNA replication licensing protein CDT1. These phenotypes, which are quite distinct to the inhibition of Aurora kinase, may all be used to specifically define the in vivo inhibitory activity of CBB2001, or other Plk1 inhibitory chemicals. In addition, mitotic inhibition of Plk1 by CBB2001 delays the exit of mitosis, which was revealed by delayed removal of APC/C substrates, such as Geminin, Cyclin B1 and Aurora A. Although CBB2001 shows good inhibitory activity in various tumor cells, it exerts less toxic effects on normal cells. Finally, we confirmed the tumor inhibitory activity of CBB2001 in tumor xenograft models, indicating its promising potential to be anti-cancer chemicals.
MATERIALS AND METHODS
Cell Culture, Synchronization and Microscopy
Human lung carcinoma A549, cervical carcinoma HeLa, and transformed embryonic kidney 293T cells, mouse teratocarcinoma F9 cells, mouse embryonic fibroblasts (MEF), and normal liver NCTC1469 and Chang cells were grown in DMEM; human colorectal carcinoma HCT116, rhabdoid tumor G401 and osteosarcoma U2OS were in McCoy’s5a; human ovarian carcinoma A2780, non-small-cell lung carcinoma H1299, breast carcinoma T47D cells and normal liver QSG-7701 cells were in RPMI-1640; ovarian teratocarcinoma PA-1 and colon carcinoma RKO cells were in EMEM media; neuroblastoma SH-SY5Y cells were in ATCC complete medium (a mixture of Eagle’s Minimum Essential Medium and Ham’s F12 Medium, 1:1), respectively. All media were supplemented with 10% FBS and antibiotics. All cells lines were used according to the protocols of the cell bank, detected for protein markers as p53, Sox2 and cell morphology for validation. Tumor types and cell sources were listed in Supplementary Table S1.
HeLa cells were synchronized by thymidine (2 mM) or nocodazole (50 ng/ml) as described specifically for each experiment.25 For serum starvation (G0/G1 block),26 cells at 40% confluency were washed twice with 1 × PBS and were subsequently incubated in DMEM plus 0.1% serum for 72 h. The cells were restimulated with 10% serum for cell cycle re-entry. Cell morphology was examined and imaged using a Zeiss Axio Observer A1 microscope equipped with charged coupled device.
Plasmids and Proteins
The kinase domain of human Plk1 (36–343 a.a.), which lacks the amino terminus inhibitory region, and human Aurora A (36–403 a.a.) were fused with the histiditine6 epitope tag in pET28-a (+) vector.19 The proteins were expressed in E. coli BL21 strain and affinity purified using Ni Sepharose affinity resin (GE Healthcare, UK).
Chemicals and Antibodies
CBB2001 was designed and synthesized using thiazole as the molecular scaffold with procedures described in Supplementary Figure S1.27 ON01910 was purchased from Onconova Therapies; LFM-A13 was from Cayman Chemical; VX680 was from Chemie Teck and ZM447439 was from Santa Cruz Technologies, respectively. Calf thymus histone H1 was from Roche Diagnostics and histone H3 was from New England Biolabs. Anti-Cyclin A, CDK2, DDB1 and Geminin antibodies were described previously.28, 29, 30 The anti-Plk1 antibody was from Bethyl Laboratories, anti-Phospho-Plk1 T210 (no. 558400) antibody was from BD Pharmingen, anti-Aurora A antibody (no. 4718) was from Cell Signaling Technologies and anti-Cyclin B1(sc-245) antibody was from Santa Cruz Technologies.
Flow Cytometry Assay
Cells were trypsinized for detachment, centrifuged and rinsed in 1 × PBS twice, and then fixed in cold 70% ethanol. Before analysis, propidium iodide (10 μg/ml) and RNase A (0.1 mg/ml) were added to the cells for nucleic acid staining. Samples were analyzed in a Flow Cytometry Analyzer (Cytomic FC500, Beckman Coulter) and results were processed with Flowjo software (Treestar).
Cell Growth Inhibition Assays (MTT)
HeLa cells were exponentially grown in 96-well plates and were dosed with various concentrations of Plk1 kinase inhibitory chemicals for 24 h. They were analyzed using the MTT assay by addition of equal amounts of 3-(4, 5-dimethylthiazol-2-yl)-2 and 5-diphenyltetrazolium bromide (0.5 mg/ml) for 4 h to produce formazan in living cells. The reaction products were dissolved by addition of dimethyl sulfoxide and the absorbance of the solutions was measured in Bio-Rad iMark microplate reader. Quadruplicates were performed for each dose of the chemicals. Data procession and graphing (such as IC50 or EC50 calculation) were managed with the GraphPad Prism 5 software.
Small RNA Interferences
The human Plk1 siRNAs, AAGGGCGGCTTTGCCAAGTGCTT and AACCAGUGGUUCGAGAGACAG, were described previously 31, 18 The Plk1 shRNA, CGGCAGCGTGCAGATCAACTT, was constructed into the pSuper vector and used as previously described.32
In Vitro Kinase Assays
Recombinant Plk1 or Aurora A kinase protein (0.5 μg) was incubated with or without various Plk1 or Aurora kinase inhibitors in the kinase buffer (50 mM MOPS, pH 7.5, 10 mM MgCl2, 0.1 mM Na3VO4, 5 mM beta-glycerophosphate, 2 mM DTT), 10 μM ATP and 5 μCi [γ32P]-ATP (10 mCi/ml, Beijing Furui Bioengineering), using 1 μg casein for Plk1 or Histone H1 or H3 for Aurora A as substrates. The reactions were incubated at 30°C for 30 min,33 and the phosphorylated proteins were separated in SDS-PAGE and visualized by autoradiography. Alternatively, kinase activities were measured by using the Kinase-Glo Max Luminescent Kinase Assay Kit (Promega, V6071), using peptide DDLLEDSFADEDE according to the manufacturer’s protocols.34
Isothermal Titration Calorimetry
ITC experiments were performed with the MicroCal iTC200 titration calorimeter (Northampton, MA). Plk1 kinase domain proteins (15 μM) and CBB2001 (250 μM) were preserved in 50 mM Tris at pH 7.4, 500 mM NaCl and 1 mM DTT, 1% DMSO. Titrations were carried out at 25°C with a stirring speed of 300 r.p.m. and a 120-s duration between each 2 μl injection. Parallel experiments were performed by injecting CBB2001 into the buffer to determine heats of dilution, which were subtracted from their respective titrations in the final figure.
Mouse Xenograft Models
Athymic nude mice (BALB/cASlac-nu) were obtained from Shanghai Slac Laboratory Animal, Chinese Academy of Sciences, with licensing number of SCXK-2007-0005. Mice were maintained in isolated bio-safety facility for specific pathogen free (SPF) animals. All operations were carried out in accordance with the National Standard of Animal Care and Use Procedures (20080820). For tumor suppression assays using the mouse xenograft model,35 actively growing human HeLa or A2780 cells were trypsinized, harvested and resuspended in DMEM or RPMI-1640, respectively. 1 × 106 cells in 100 μl volume were injected s.c. into the flanks of female athymic nude mice (5 weeks old). After 10–15 days when the tumor volume was about 100–150 mm3, animals were randomized into groups of five for drug studies. CBB2001 (25 mg/kg) was i.p. injected once every 2 days starting on day 0. Tumor volumes were measured by calipers (accuracy of 0.02 mm) every other day and calculated using the equation V=(L × W2)/2, in which L and W refer to the larger and smaller dimensions. Each tumor was independently measured and calculated by changes in volume (folds) relative to day 0. Statistical significances between groups were tested by one-way analysis of variance.
RESULTS
CBB2001 Efficiently Inhibits the Kinase Activity of Plk1
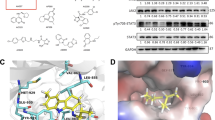
To examine the function and regulation of Plk1, we developed a new thiazole derived chemical named CBB2001, an ATP analog, which is designed to properly dock into the kinase-pocket of Plk127, 36, 37 (Figures 1A and B, Supplementary Figure S2A). CBB2001 docks to the hinge region of ATP binding pocket of Plk1, through two hydrogen bonds from benzimidazole to the NH backone of Cys 133 and the amino group of the benzene ring to the main chain carbonyl of Leu 59. These interactions place the benzimidazole and thiazole groups of CBB2001 in the adenine and ribose portion of the ATP pocket in Plk1. Binding affinity is further enhanced by two π–π stacking interactions. One is between Phe 183 and the benzimidazole group of CBB2001, and the other is between Phe 183 and the chlorophenyl group of CBB2001. Calculation of the docking model showed a binding free energy of ΔG=−9.7 kcal/mol. The synthetic routes of CBB2001 were shown in Supplementary Figure S1.38 The isothermal titrations were performed to measure the binding energy and a value of −6.70 kcal/mol was obtained (Supplementary Figure S2C). As Plk1 kinase domain is very flexible and easy to precipitate,39 the measurement confirmed that CBB2001 is a novel small molecule that interacts with the kinase domain of Plk1 with high affinity.

The new compound CBB2001 efficiently inhibits the Plk1 kinase activity in vitro. (a) Chemical structures of newly developed Plk1 kinase inhibitory chemical CBB2001, and two reported Plk1 inhibitors ON01910 and LFM-A13. (b) Auto-docked binding model of CBB2001 in Plk1. (A) Ribbon structure display of CBB2001 binding to active sites of Plk1 (PDB code: 2owb; ref.36). CBB2001 is shown in yellow and protein residues involved in CBB2001 binding are highlighted in cyan. (B) Surface display of CBB2001 (yellow sticks) in the Plk1 kinase-pocket. The molecular docking and binding affinity (ΔG) was calculated by AutoDockTools-1.5.4 and AutoDock Vina-1.1.2. Images were prepared by using PyMOL (DeLano Scientific LLC). Hydrogen bonds were indicated with dashed lines. (c) In vitro kinase assay for Plk1 inhibitors. Purified Plk1 kinase protein was incubated with 1 μg of casein (substrate) and chemical inhibitors with concentrations as indicated. The 32P-labeled casein was SDS-PAGE separated and visualized by autoradiography. (d) The half maximal inhibitory concentrations IC50 of CBB2001 in vitro kinase activity on Plk1 as well as Aurora and CDK2 were tabulated. (e) In vitro kinase assay for Aurora kinase inhibitors using the purified Aurora A kinase and histone H3 or H1 as substrates as indicated.
We analyzed the in vitro inhibitory effects of CBB2001 on Plk1 kinase, as well as that of other two reported Plk1 inhibitors, ON01910, a non-ATP competitive compound, and LFM-A13, a leflunomide metabolite analog (Figure 1A). We found that CBB2001 completely inhibited the Plk1 kinase activity at 0.8 μM, with an IC50 of 0.39 μM, while LFM-A13 showed slight inhibition at 10 μM, whereas ON01910 did not have any detectable inhibitory effects on Plk1 under the same conditions (Figure 1C). These data indicate that CBB2001 exhibits more effective inhibition on Plk1 in vitro than that of LFM-A13 and ON01910.
As CBB2001 is designed as an ATP analog and the concentration of ATP in living cells is in the millimolar range, we carried out an ATP competition assay to test the affinity of CBB2001 towards Plk1. As shown in Figure 1D, CBB2001 displayed a high affinity towards Plk1 with an IC50 of 0.25 μM in the presence of 0.5 mM ATP. Even in the presence of 1 mM ATP, the inhibitory activity of CBB2001 was only reduced less than two-folds, with an IC50 of 0.4 μM. These studies suggest that CBB2001 is an effective Plk1 inhibitor that targets at the kinase domain of Plk1 (Figure 1D and Supplementary Figure S3A). Because of the structural similarity of the kinase-pocket between mitotic kinases, we tested whether CBB2001 has any inhibitory effects on other two mitotic kinases, Aurora A and CDK2. We found that CBB2001 did not show detectable inhibitory activity on the recombinant Aurora A kinase, even at 10 μM, using either histone H1 and H3 as substrates, while Aurora kinase inhibitor VX680 can inhibit the activity of Aurora A kinase at 1–2 μM (Figure 1E). Conversely, our analyses also showed that Aurora kinase inhibitors VX680 and ZM447439 did not have any detectable inhibitory activities towards Plk1 (Figure 1C). Further analyses revealed that CBB2001 has minimum effects on both Aurora A kinase and CDK2, with an IC50 of 85.2 μM for Aurora A and 150.5 μM for CDK2, respectively, (Figures 1D, E and Supplementary Figure S3B). Our studies thus indicate that CBB2001 is a specific chemical inhibitor for Plk1, which displays a better inhibitory activity than that of previously reported Plk1 inhibitors, LFM-A13 and ON01910.
CBB2001 Induces Cell Cycle Arrest and Formation of Mitotic ‘Polo’ Cells in Cancer Cells
To analyze the effects of the new Plk1 inhibitor in cancer cells, HeLa cells were initially treated with increasing concentrations of CBB2001 for 16 h. This treatment led to the increased mitotic arrest with many cells displaying a mitotic ‘polo’ phenotype, even at 0.5 μM of CBB2001 (Figure 2a and Supplementary Figure S4). Flow cytometry analysis of CBB2001 treated cells also indicated that the new Plk1 kinase inhibitory chemical caused a cell cycle arrest with the cells accumulated in the G2/M phases with 4 N DNA content. Ablation of Plk1 expression with specific siRNAs or shRNAs phenocopied the mitotic cell cycle arrest and mitotic ‘polo’ phenotypes (Supplementary Figure S5), suggesting CBB2001 specifically inhibits Plk1 in HeLa cells. However, although ON01910 and LFM-A13 have been used as PLK1 inhibitors, we found ON01910 can induce the similar mitotic ‘polo’ cells only at much higher concentrations (>5 μM) while LFM-A13 did not produce any ‘polo’ effect even at 10 μM (Figure 2b). Quantitative analyses indicate that CBB2001 is more specific and effective to inhibit Plk1 activity in vivo to induce mitotic cell cycle arrest with the ‘polo’ cells phenotypes (EC50 of 0.32 μM), as compared with that of ON01910 (EC50=5.75 μM) in HeLa cells (Figure 2c).

CBB2001 effectively induces mitotic arrest of cancer cells with mitotic ‘polo’ phenotypes. (a), (b) HeLa cells were treated with CBB2001 as well as reported Plk1 inhibitory chemicals ON01910 and LFM-A13, for 20 h. They were analyzed by microscopy and flow cytometry. Bar=40 μm. (c) The percentage induction of the mitotic ‘polo’ cells for experiments in (a) or (b) was used to calculate the EC50 of CBB2001 or ON01910, respectively.
As Aurora kinases were reported to phosphorylate Plk1 at threonine 210 (T210) and such a modification activates the Plk1 kinase activity, we also examined the effects of various concentrations of Aurora kinase inhibitors VX680 and ZM447439 in HeLa cells. Although inhibition of Aurora kinases by VX680 or ZM447439 was sufficient to arrest cells in the G2 phase at low concentrations (Supplementary Figure S6), these Aurora kinase inhibitors induced much less ‘polo’ cells, as compared with that of CBB2001. Our analysis indicates that while CBB2001 is specific for Plk1 inhibition in vivo, the activity of Plk1 may be partially affected by the Aurora kinase inhibitors. The differences in the phenotypes caused by Plk1 and Aurora kinase inhibitors suggest that these two kinases have temporally distinct functions in the cell cycle.
CBB2001 Inhibits the Growth of Various Cancer Cells and Prevents Tumor Growth in Mouse Xenograft Models
Plk1 has been shown to be an excellent target in cancer therapy as it is overexpressed in a variety of human cancers, which is often associated with poor prognosis and low overall survival. To further evaluate the application of CBB2001 as a potential anti-cancer agent, we examined whether the new Plk1 inhibitor can block the growth of cancer cells derived from other types of cancers. A panel of cancer cell lines derived from different types of human or mouse cancers, including ovarian, breast and lung carcinomas as listed in Supplementary Table S1, were tested to examine the growth inhibitory activities of CBB2001 (Figure 3). Our studies revealed that CBB2001 caused the G2/M cell arrest associated with the mitotic ‘polo’ phenotype in all of these cancer cells (Figures 3 and 4d) while LFM-A13 or ON01910 either exhibited no effects or required much higher concentrations (Figure 2b), respectively. For example, CBB2001 suppressed the growth of ovarian carcinoma A2780 cells with an IC50 of 0.43 μM; teratocarcinoma F9 cells, 0.37 μM; and HeLa cells, 0.85 μM; while the parallel tests of LFM-A13 or ON01910 showed the IC50 of 37.36 or 6.46 μM, respectively (Figures 4d and 5a, Supplementary Table S2). To determine whether CBB2001 has similar effects on normal non-transformed cells, we tested the effects of CBB2001 on human normal hepatic QSG-7701 cells and MEF (Figures 4a–c). We found that both QSG-7701 and MEF were less sensitive to CBB2001 than HeLa cells for the induction of the G2/M-phase arrest and the mitotic ‘polo’ cells (Figure 4c). We also examined the growth inhibition effects of CBB2001 on additional human normal hepatocyte QSG-7701 and Chang liver cells, as well as mouse normal liver NCTC1469 cells by using MTT assays. All of these normal cells showed less sensitivity to CBB2001, with 5–8 folds of higher IC50 for cell growth inhibition than that of cancer cells (Figure 4d and Supplementary Table S2). These studies suggest that non-transformed cells are less sensitive to CBB2001.

CBB2001 induced the G2/M cell cycle arrest and the mitotic ‘polo’ phenotype in many types of cancer cells. Indicated cancer cell lines were each dosed with CBB2001 (1 μM) or DMSO (control) for 20 h. Cells were collected and analyzed by microscopy and flow cytometry.

Plk1 inhibition by CBB2001 suppresses growth of cancer cells but normal cells are less sensitive. (a), (b) Human normal liver QSG-7701 cells and mouse normal diploid mouse embryonic fibroblasts (MEF) were treated with CBB2001 for 20 h. The effects of CBB2001 were analyzed by microscopy and flow cytometry. (c) CBB2001 induced the G2 phase arrest and the formation of mitotic ‘polo’ cells in HeLa cells, but had less effects on normal liver QSG-7701 or MEF cells. (d) Human normal liver QSG-7701 and Chang cells, as well as mouse normal liver NCTC1469 cells, showed less sensitivity to the growth inhibition by CBB2001, as compared with that of human ovarian cancer A2780 or mouse teratocarcinoma F9 cells.

CBB2001 inhibits tumor growth in cultured cells and in mouse xenograft tumor models. (a) Human cervical cancer HeLa cells were maintained in 96-well plates and treated with Plk1 inhibitory chemicals CBB2001, ON01910 and LFM-A13 for 20 h. They were analyzed using MTT cell growth inhibition assays to calculate IC50. Quadruplicates were performed. CBB2001 showed better inhibition activity towards HeLa cells than that of ON01910 or LFM-A13. (b) CBB2001 inhibited xenograft tumor growth of HeLa and A2780 cells in the mouse. Mouse xenograft models were established as described in ‘MATERIALS AND METHODS’. The animals were treated with CBB2001 (25 mg/kg) or vehicle (DMSO) as a control. Tumor volumes were measured and calculated for fold change. Differences between groups were compared by one-way analysis of variance (*P<0.05; **P<0.01).
To assess whether CBB2001 exhibits anti-tumor activities in animals, we further examined the effects of CBB2001 on the mouse tumor xenograft models. Although inoculation of HeLa or A2780 cells induced tumor formation in the immunodeficient (nude) mice (Figure 5b), the tumors in both xenograft models show remarkably growth inhibition after the treatment of CBB2001 (25 mg/kg), but not vehicle control (Figure 5b), indicating the potential use of CBB2001 as an anti-tumor chemical through the inhibition of Plk1.
CBB2001 Induces Accumulation of Plk1 and Geminin Proteins in Mitotic Arrested Cells
To further analyze the effects of Plk1 inhibitors at the molecular level, we monitored several cell cycle related proteins in their response to Plk1 inhibitors. Treatment with increasing concentrations of CBB2001 from 0.001 to 1 μM (Figure 6a) led to the gradual accumulation of total Plk1 protein and a modified Geminin protein (Figures 6b and c), which migrates in SDS-PAGE protein gel at a slower mobility than that of the majority of Geminin. However, ON01910 and LFM-A13 did not induce similar accumulation of Plk1 protein and modified Geminin at these concentrations (Figure 6b). Consistent with the notion that CBB2001 specifically inhibited Plk1 activity in vivo and in vitro, ablation of Plk1 expression by small RNA interferences also led to the accumulation of the modified Geminin protein (Supplementary Figure S5A). Thus, our studies revealed that the accumulation of Plk1 and modified Geminin proteins may serve as molecular indicators for the cellular inhibition of Plk1. Using these sensitive assays, CBB2001 showed an EC50 (Half Maximal Effective Concentration) of 0.43 μM for the appearance of the modified Geminin or 0.61 μM for Plk1 protein accumulation (Figure 6c).

Accumulation of Geminin and Plk1 protein in mitotic arerested ‘polo’ cells after CBB2001 treatment. (a) Treatment of asynchronously growing HeLa cells by various Plk1 inhibitors for 14 h. Morphological images of the cells were taken with a Zeiss Axio Observer A1 microscope with × 10 objective lens. Bar=40 μM. CBB2001 induced mitotic arrest cells with ‘polo’ phenotype at 1 μM. (b) HeLa cells treated with various concentrations of Plk1 inhibitors as described in (a) were harvested and western blotting analyzed for the indicated proteins using specific antibodies. Geminin and Plk1 protein were accumulated in mitotic arrest and polo emerged cells induced by CBB2001 at 1 μM. Samples treated with Aurora kinase inhibitors VX680 (VX) or ZM447439 (ZM) were also blotted. (c) Geminin and Plk1 protein accumulation were used to quantify the in vivo effect of CBB2001 Plk1 inhibitory activities. The half maximal effective concentrations (EC50) were calculated using GraphPad Prism 5 software, respectively.
Aurora Kinase Inhibitors Do Not Phenocopy the Inhibition of Plk1 Activity in Mitosis
Aurora kinases have been shown to phosphorylate T210 in Plk1, which can be monitored by a Plk1 phospho-T210-specific antibody, during the cell cycle progression from the G2 phase to mitosis, leading to the activation of the kinase activity of Plk1. In our experiments, we found that while inhibition of both Plk1 and Aurora kinases caused the G2/M cell cycle arrest, the percentages of mitotic ‘polo’ cells induced by Aurora kinase inhibitors appeared to be obviously less than that of our new Plk1 inhibitor CBB2001. Using the accumulation of Plk1 and the modified Geminin proteins as a marker for Plk1 inactivation by CBB2001 and Plk1-specific siRNA/shRNA, treatment of HeLa cells with Aurora kinase inhibitors VX680 and ZM447439 did not cause the accumulation of both proteins (Figure 6b), suggesting that Aurora kinase inhibitors did not arrest cancer cells at the same cell cycle stages as that of inactivation of Plk1 even through Aurora kinases can activate Plk1. To further analyze the regulation of Plk1 phosphorylation at T210 at different stages during mitosis, HeLa cells were first synchronized by the double thymidine block to arrest cells at the G1/S border.25 As the synchronized cells were released into fresh medium, the cells were treated with nocodazole for 16 h to allow them to progress through S, G2, and to arrest the cell cycle in mid-mitosis (meta-phase). To monitor the effects of CBB2001 and Aurora kinase inhibitors, we added these kinase inhibitors in the last 4 h during the nocodazole treatment.25 Surprisingly, we found that treatment of nocodazole-arrested mtiotic cells with Aurora kinase inhibitors VX680 and ZM447439 at 1 and 2 μM, which effectively inhibit Aurora kinase activity (Figure 1E), did not affect the phosphorylation status of Plk1 at T210 (Figure 7a). Similar results were obtained when the nocodazole-treated mitotic cells were shaken off and replated into nocodazole-containing to ensure the cells are in meta-phase (Figure 7b). In addition, while the mitotic modified and accumulated form of Geminin is insensitive to Plk1 inhibitors CBB2001, ON01910 or LFM-A13, it decreased after the treatment with Aurora kinase inhibitor VX680 (Figures 7a and b), suggesting that Aurora kinase inhibitors are still active but they do not affect the T210 phosphorylation in Plk1 while the cells are still arrested in mid-mitosis.

The phosphorylation of T210 on Plk1 in mitotic cells is insensitive to Aurora kinase inhibitors. (a) Effects of Plk1 and Aurora kinase inhibitors on synchronized G1/S cells progressing into mitosis. HeLa cells were synchronized in the G1/S border by 2 mM thymidine for 16 h and then released from thymidine block into fresh medium containing nocodazole (50 ng/ml). The cells were incubated for additional 16 h to allow them to progress into mitosis and to be arrested in mitosis. In the last 4 h during nocodazole treatment, various concentrations of Plk1 and Aurora kinase inhibitors were added as indicated. The proteins were analyzed by western blotting with specific antibodies. (b) HeLa cells were arrested in mitosis by nocodazole (50 ng/ml) for 16 h. The mitotic polo cells were shaken off and replated into fresh medium containing nocodazole (50 ng/ml). The cells were incubated with various concentrations of Plk1 and Aurora kinase inhibitors for 4 h in the presence of nocodazole as indicated. Proteins samples were collected and analyzed as in (a). (c) Sensitivity of T210 phosphorylation of Plk1 in synchronized HeLa cells progressing from S to G2 phases. The G1/S cells arrested by thymidine block or serum starvation were released into fresh medium and incubated with CBB2001 or VX680 at various concentrations for 12 h. Proteins were analyzed by western blotting with specific antibodies.
To further investigate the effects of CBB2001 and Aurora kinase inhibitors, the G1/S arrested cells or serum-starved cells were released into fresh media containing CBB2001 or VX680. The cells were allowed to progress through S and G2 phases in the presence of inhibitors for 12 h. In this case, the phosphorylation of Plk1 at T210 is inhibited by VX680, but this inhibitory effect of VX680 was incomplete (Figure 7c), even when the concentrations of VX680 were 1–2 μM, which are sufficient to inhibit Aurora kinases in the in vitro assay (Figure 1E). These studies suggest that Aurora kinases may only partially be responsible for the phosphorylation of T210 and the activation of Plk1 during the S phase progression into G2/M but not in nocodazole-arrested mitotic cells. Interestingly, we found that treatment of CBB2001 up to 2 μM can inhibit the phosphorylation of T210 in Plk1, raising the possibility that Plk1 itself or other kinases, may phosphorylate T210. However, further experiments are required to verify this possibility.
Inhibition of Plk1 by CBB2001 Delays Mitotic Exit
It was reported that Plk1 is required for the activation of the APC/C ubiquitin E3 ligase complex.1, 2, 3 To further analyze the regulation of Plk1 using the Plk1 inhibitory chemicals during mid-mitosis to the exit of mitosis, we monitored several reported APC/C substrates. The nocodazole-arrested mitotic cells were removed by mitotic shake-off, washed and replated into fresh culture medium. This process allows the mitotic arrested cells to finish mitosis and to progress towards the exit of mitosis, which is associated with the degradation of many APC/C substrates.40, 41 Consistently, we found that the removal of nocodazole from the mitotic arrested cells led to the proteolysis of APC/C substrates Geminin, Cyclin B1 and Aurora A in a time-dependent process. Treatment of Plk1 inhibitor CBB2001 can significantly delay the degradation of Geminin, Cyclin B1 and Aurora A during the mitotic exit, suggesting that Plk1 is required for the exit of mitosis from mitotic arrested cells (Figure 8). The removal of Geminin, Cyclin B1 and Aurora A can be monitored by western blotting and quantified by the grayscale density of protein bands (Figure 8c). In cells that are exiting mitosis, there are again no significant effects on the phosphorylation of T210 in Plk1 after treatment of Aurora kinase inhibitors (Supplementary Figure S7). Aurora kinase inhibitors also did not have significant effects on the degradation of APC/C substrates (Supplementary Figure S7). These results indicate that there is substantial difference in the inhibitory effects of Plk1 and Aurora kinase inhibitors during the cell cycle progression from mitosis to the exit of mitosis.

CBB2001 delays the exit of mitosis by inhibition of Plk1. (a, b) HeLa cells were arrested in mitosis by sequential treatment of thymidine (2 mM) and nocodazole (50 ng/ml) as described in Figure 6a. The mitotic cells were shaken off and replated into fresh medium without nocodazole but with DMSO (control), or with 1 μM of CBB2001 (a) or other Plk1 inhibitors ON01910 (10 μM) and LFM-A13 (10 μM) (b) as indicated. The cells were harvested at various time points after nocodazole release and the proteins were analyzed by western blotting using specific antibodies. (c) Efficiency of Plk1 kinase inhibitory chemicals on mitotic exit was quantified by protein removal of Geminin, Cyclin B1 and Aurora A in cells as experiments in (a), (b). Protein levels were characterized by grayscale density of bands and transferred to normalized ratio for graphing.
DISCUSSION
Plk1 is a key kinase that regulates centrosome maturation, spindle assembly, chromosome condensation and segregation, and cytokinesis. It is overexpressed in many cancers and serves as a target for cancer therapy. In addition, the unique kinase structure of Plk1 and its regulatory mechanisms provide the molecular basis for designing various chemical inhibitors. Both ATP analogs and non-ATP compounds have been developed as Plk1 kinase inhibitors that were used to dissect its function in the cell cycle or serve as potential anti-cancer chemicals in clinical trials.33
In this study, we have developed a new Plk1 inhibitor, CBB2001, to explore the regulation of mitosis by Plk1 and cellular phenotypes of Plk1 inhibition. Our results indicate that CBB2001 displays an excellent inhibitory effect on Plk1 kinase activity both in vitro and in vivo, compared with that of other reported Plk1 inhibitors such as LFM-A13 or ON01910. CBB2001 inhibits Plk1 activity in cultured cells and induces mitotic ‘polo’ phenotype of cells, as well as the accumulation of Plk1 and Geminin proteins, which are distinct from that of the inhibition of Aurora kinases. Our studies suggest that the accumulation of Plk1 and Geminin proteins can be used to quantitatively characterize the inhibitory activity of CBB2001 to Plk1 in vivo. Moreover, mitotic inhibition of Plk1 mediated by CBB2001 delays the mitosis exit. Interestingly, the degradation of Geminin, one of the APC/C substrates, is delayed by mitotic inhibition of Plk1, which may serve as an excellent biomaker for PLK1 inhibition by CBB2001, as well as other Plk1 inhibitory chemicals.
Most remarkably, our studies suggest that the phosphorylation of T210 and Plk1 activation in mid-to-late mitosis and during mitotic exit appears to be insensitive to Aurora kinase inhibitors. As Aurora kinases have been shown to phosphorylate T210 in Plk1 during the cell cycle progression from the G2 phase to mitosis, our evidence suggests that additional regulatory mechanisms exist in cells in mid- and later mitosis to maintain the phosphorylation status of T210 and the activity of Plk1. As the expression of both Plk1 and Aurora kinases are altered in many human cancers, our work suggests that Plk1 and Aurora kinases serve as excellent and independent targets for cancer therapy.
References
Strebhardt K, Ullrich A . Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 2006;6:321–330.
Barr FA, Sillje HH, Nigg EA . Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 2004;5:429–440.
Eckerdt F, Strebhardt K . Polo-like kinase 1: target and regulator of anaphase-promoting complex/cyclosome-dependent proteolysis. Cancer Res 2006;66:6895–6898.
Moshe Y, Boulaire J, Pagano M et al. Role of Polo-like kinase in the degradation of early mitotic inhibitor 1, a regulator of the anaphase promoting complex/cyclosome. Proc Natl Acad Sci USA 2004;101:7937–7942.
Nakajima H, Toyoshima-Morimoto F, Taniguchi E et al. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J Biol Chem 2003;278:25277–25280.
Toyoshima-Morimoto F, Taniguchi E, Nishida E . Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep 2002;3:341–348.
Golsteyn RM, Mundt KE, Fry AM et al. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol 1995;129:1617–1628.
Oshimori N, Ohsugi M, Yamamoto T . The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat Cell Biol 2006;8:1095–1101.
Baumann C, Korner R, Hofmann K et al. PICH a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 2007;128:101–114.
Pouwels J, Kukkonen AM, Lan W et al. Shugoshin 1 plays a central role in kinetochore assembly and is required for kinetochore targeting of Plk1. Cell Cycle 2007;6:1579–1585.
Kurasawa Y, Yu-Lee LY . PICH and cotargeted Plk1 coordinately maintain prometaphase chromosome arm architecture. Mol Biol Cell 2010;21:1188–1199.
Wu ZQ, Liu X . Role for Plk1 phosphorylation of Hbo1 in regulation of replication licensing. Proc Natl Acad Sci USA 2008;105:1919–1924.
Takahashi T, Sano B, Nagata T et al. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci 2003;94:148–152.
Yamamoto Y, Matsuyama H, Kawauchi S et al. Overexpression of polo-like kinase 1 (PLK1) and chromosomal instability in bladder cancer. Oncology 2006;70:231–237.
Schoffski P . Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist 2009;14:559–570.
Kanaji S, Saito H, Tsujitani S et al. Expression of polo-like kinase 1 (PLK1) protein predicts the survival of patients with gastric carcinoma. Oncology 2006;70:126–133.
Liu X, Erikson RL . Activation of Cdc2/cyclin B and inhibition of centrosome amplification in cells depleted of Plk1 by siRNA. Proc Natl Acad Sci USA 2002;99:8672–8676.
Guan R, Tapang P, Leverson JD et al. Small interfering RNA-mediated Polo-like kinase 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals. Cancer Res 2005;65:2698–2704.
Macurek L, Lindqvist A, Lim D et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008;455:119–123.
Seki A, Coppinger JA, Jang CY et al. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008;320:1655–1658.
Keen N, Taylor S . Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer 2004;4:927–936.
Taylor S, Peters JM . Polo and Aurora kinases: lessons derived from chemical biology. Curr Opin Cell Biol 2008;20:77–84.
Meraldi P, Honda R, Nigg EA . Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J 2002;21:483–492.
Bischoff JR, Anderson L, Zhu Y et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J 1998;17:3052–3065.
Lu F, Lan R, Zhang H et al. Geminin is partially localized to the centrosome and plays a role in proper centrosome duplication. Biol Cell 2009;101:273–285.
Cooper S, Iyer G, Tarquini M et al. Nocodazole does not synchronize cells: implications for cell-cycle control and whole-culture synchronization. Cell Tissue Res 2006;324:237–242.
Emmitte KA, Adjabeng GM, Andrews CW et al. Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding. Bioorg Med Chem Lett 2009;19:1694–1697.
Higa LA, Banks D, Wu M et al. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle 2006;5:1675–1680.
Higa LA, Mihaylov IS, Banks DP et al. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol 2003;5:1008–1015.
Higa LA, Wu M, Ye T et al. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol 2006;8:1277–1283.
Liu X, Erikson RL . Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci USA 2003;100:5789–5794.
Chen Q, Zhang X, Jiang Q et al. Cyclin B1 is localized to unattached kinetochores and contributes to efficient microtubule attachment and proper chromosome alignment during mitosis. Cell Res 2008;18:268–280.
Lenart P, Petronczki M, Steegmaier M et al. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol 2007;17:304–315.
Johnson EF, Stewart KD, Woods KW et al. Pharmacological and functional comparison of the polo-like kinase family: insight into inhibitor and substrate specificity. Biochemistry 2007;46:9551–9563.
Morton CL, Houghton PJ . Establishment of human tumor xenografts in immunodeficient mice. Nat Protoc 2007;2:247–250.
Trott O, Olson AJ . AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–461.
Kothe M, Kohls D, Low S et al. Structure of the catalytic domain of human polo-like kinase 1. Biochemistry 2007;46:5960–5971.
Zhong H, Xin S, Zhao Y et al. Genetic approach to evaluate specificity of small molecule drug candidates inhibiting PLK1 using zebrafish. Mol Biosyst; 6:1463–1468.
Yun SM, Moulaei T, Lim D et al. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat Struct Mol Biol 2009;16:876–882.
Bashir T, Dorrello NV, Amador V et al. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 2004;428:190–193.
Wei W, Ayad NG, Wan Y et al. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004;428:194–198.
Acknowledgements
We appreciate the assistance from lab members for their kind suggestions and advice. We also thank Professor Xiaoning Wang and Dr Yuhui Chen for kindly providing Chang liver cells (School of Bioscience and Bioengineering, South China University of Technology), to Drs Song Li and Shengchang Xin for the use of SPF animal bio-secure facility and providing the standard operating procedures of animal experiments, and to Ms Zhuozhu for technical support and advice for the isothermal titration calorimetry experiments. This work was supported by the grants from National Natural Science Foundation of China (NSFC) (30971616 and 21133002), and Science, Industry, Trade and Information Technology Commission of Shenzhen Municipality (SZSITIC) (Biology, Internet, New Energy Industries Development Special Funds for Basic Research Projects of Shenzhen) (JC201104210125A), and the Pfizer (China)-PKU collaboration program. The funders had no role in project design, experiments performance, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Polo-like kinase 1 (Plk1) is a serine/threonine kinase that has a critical role in mitosis. Plk1 expression is associated with cell proliferation and is overexpressed in a variety of cancers. A novel kinase inhibitory chemical, CBB2001, is described that specifically inhibits Plk1 kinase activity, indicating its potential as an anti-cancer agent.
Supplementary information
Rights and permissions
About this article
Cite this article
Lan, R., Lin, G., Yin, F. et al. Dissecting the phenotypes of Plk1 inhibition in cancer cells using novel kinase inhibitory chemical CBB2001. Lab Invest 92, 1503–1514 (2012). https://doi.org/10.1038/labinvest.2012.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2012.114
