
Abstract
Phosphatidylserine (PS)-containing liposomes (PSLs) strongly inhibit inflammatory bone loss in adjuvant arthritic (AA) rats. This effect was attributed to the inhibition of osteoclastogenesis through the secretion of prostaglandin E2 and transforming growth factor-β1 by osteoclast precursors after the phagocytosis of PSLs. However, infiltrated macrophages are considered to secrete anti-inflammatory mediators after phagocytosis of PSLs, which also contribute to inhibiting osteoclastogenesis. In the present study, we have attempted to elucidate the effects of PSLs on the phenotype of infiltrated macrophages during inflammatory bone loss. In AA rats, the ankle joints swelled with the infiltration of both macrophages and helper T cells into the synovium after a complete Freund's adjuvant injection. In the ankle joints of AA rats, approximately half of the infiltrated macrophages underwent a phenotypic change from interleukin (IL)-1β-producing to IL-10-producing cells after the phagocytosis of PSLs. In lipopolysaccharide (LPS)-stimulated macrophages, PSLs also significantly decreased IL-1β production, but increased IL-10 production. Moreover, PSLs inhibited the rapid activation of p38 mitogen-activated protein kinases (MAPK) and nuclear factor (NF)-κB, but enhanced the delayed activation of extracellular signal-regulated kinase (ERK) in LPS-stimulated macrophages. PSL-induced different influence on the activities of p38 MAPK and ERK is a likely underlying mechanism for phenotypic change of infiltrated macrophages after the phagocytosis of PSLs. This phenotypic change may be responsible for a significant decrease in the mean mRNA level of the receptor activator of NF-κB (RANK) and the RANK ligand (RANKL) in the ankle joint of PSL-treated AA rats, resulting in the inhibition of inflammatory bone loss.
Similar content being viewed by others


Main
Apoptosis is known to be associated with the resolution of inflammation by regulating the release of cytokines.1, 2, 3, 4 Accumulating evidence shows that exposure to phosphatidylserine (PS) has a central role in the recognition and phagocytosis of apoptotic cells by phagocytes.5, 6, 7 PS-dependent phagocytosis of apoptotic cells is believed to trigger the secretion of anti-inflammatory cytokines and restrains that of pro-inflammatory cytokines from macrophages8, 9 and dendritic cells.10, 11 Recently, we and others have found that PS-containing liposomes (PSLs) mimic apoptotic cells to promote the secretion of anti-inflammatory mediators, including transforming growth factor-β1(TGF-β1) and prostaglandin E2 (PGE2) by macrophages, microglia and myeloid dendritic cells in vitro.12, 13, 14 Furthermore, PSLs can promote the resolution of inflammation in vivo.2, 15, 16
More recently, we have demonstrated that a strong inhibitory effect on the inflammatory bone loss of PSLs is attributable to the inhibition of osteoclastogenesis through the secretion of both PGE2 and TGF-β1 by osteoclast precursors after phagocytosis of PSLs.17 However, it is also reasonable that infiltrated macrophages can undergo a phenotypic change from pro- to anti-inflammatory cells after phagocytosis of PSLs. This phenotypic change likely has important consequences on inflammatory bone loss, because pro-inflammatory cytokines are strong inducers of the receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL) and RANK, which have essential roles in differentiation and the activity of osteoclasts.18
Among pro-inflammatory cytokines, interleukin (IL)-1β is a master regulator of inflammation and inflammatory bone loss, because it can induce other pro-inflammatory cytokines.19, 20 Recently, much attention has been paid to IL-17 as a crucial cytokine for inflammatory osteoclastic bone loss because of its marked induction of RANKL.21 Indeed, high levels of IL-1β and IL-17 are found in the plasma and synovial tissues of rheumatoid arthritis (RA) patients, and combined blockade of IL-1β and IL-17 receptors significantly inhibited the rheumatoid arthritic osteoporosis.22 Among anti-inflammatory mediators, including TGF-β1 and PGE2, IL-10 is known as the strongest feedback mediator of inflammatory bone loss.23, 24
The paw volume increase of adjuvant arthritic (AA) rat, an animal model of RA,25, 26, 27 occurs in two stages: namely an acute inflammation stage from 10 to 21 days after complete Freund's adjuvant (CFA) injection, and the chronic bone/joint destruction stage thereafter. Therefore, in our previous study, to examine the direct effect of PSLs on osteoclastogenesis, the treatment of PSLs was started prior to the formation of osteoclasts in AA rats (14 days after CFA injection).17 However, it is not clear whether PSLs can directly influence the infiltration of macrophages and T cells. Therefore, in this study, to elucidate the direct effect of PSLs on the infiltration of macrophages and T cells, the PSLs treatment was initiated at the beginning of their infiltration in the ankle joints (10 days after CFA injection). We attempted to elucidate the effects of PSLs on the phenotype of infiltrated macrophages and T cells in the skeletal tissues of ankle joints of AA rats. We have herein demonstrated that infiltrated macrophages, but not T cells, underwent a phenotypic change from IL-1β-producing to IL-10-producing cells. This PSL-induced cell-specific phenotypic change of infiltrated macrophages also contributes to the inhibition of AA-induced trabecular bone loss through the inhibition of RANKL/RANK expression and the subsequent osteoclastogenesis.
MATERIALS AND METHODS
Reagents
Dulbecco's Modified Eagle Medium (DMEM) was purchased from Sigma (Saint Louis, MI, USA). Fetal bovine serum was obtained from Bio Whittaker (Walkersville, MD, USA). PS, phosphaidylcholine (PC), 4-nitrobenz-2-oxa-1, 3-diazole (NBD)-labeled PS and NBD-labeled PC were obtained from Avanti Polar Lipids (Alabaster, AL, USA). The mouse monoclonal anti-ED1 antibody was from BMA (Augest, Switzerland); goat polyclonal anti-IL-10, anti-IL-1β, anti-RANKL, anti-RANK, rabbit polyclonal anti-IL-17, anti-p-IκBα and mouse monoclonal anti-NF-κB antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal anti-p38, anti-p-p38, anti-extracellular signal-regulated kinase (anti-ERK) and anti-p-ERK antibodies were from Cell Signaling Technology (Beverly, MA, USA). Heat-inactivated Mycobacterium butyricum and mineral oil were purchased from Difco (Detroit, ML, USA), and thioglycolate was obtained from Nissui (Tokyo, Japan).
Liposomes
PSLs consisted of PC and PS at a molar ratio of 7:3, and PC liposomes (PCLs) consisted of PC only. PSLs and PCLs were prepared as described previously.13 In some experiments, NBD-labeled PS and PC were used to prepare NBD-labeled PSLs and PCLs, respectively.
Systemic PSLs Treatment for AA Rats and Samples Preparations
Female Lewis rats (5 weeks of age, n=108) were used for the in vivo experiments. The animals (n=81) were intradermally injected with CFA (1.5 mg heat-killed M. butyricum suspended in 0.15 ml mineral oil) at the base of the tail to induce adjuvant arthritis (AA) as described previously.28, 29, 30 Rats injected with mineral oil alone were termed sham rats (n=27). We intramuscularly injected PSLs (5 mg/kg/day, PSL-treated AA rats, n=27) or PCLs (5 mg/kg/day, PCL-treated AA rats, n=27) into the hind limbs from day 10 after CFA injection, when clinical symptoms, such as redness and swelling, were detected in the ankle joints.28, 29 AA rats intramuscularly injected with phosphate-buffered saline (PBS) used as a control (n=27). Rats were anesthetized with sodium pentobarbital (30 mg/kg, i.p.) at day 4, 11 or 18 after the treatment of PSLs or PCLs. After obtaining the blood samples, the rats (n=3 at each time point) were intracardially perfused with PBS, the ankle joints were removed and stored at −80°C for the real-time PCR analyses. The other animals were intracardially perfused with 2% paraformaldehyde (PFA) (n=6 at each time point) and the ankle joints were removed and immersed in the same fixative for 12 h at 4°C. After the μCT analyses, samples were decalcified in 10% EDTA for additional 3 weeks. After immersion in 30% sucrose in PBS for 2 days, samples were embedded in an optimal cutting temperature compound (Sakura Finetechnical, Tokyo, Japan). Serial coronal frozen sections (10 μm) were prepared for the histological and immunohistochemical analyses as described previously.26
Preparation of Peripheral Blood Monocytes
The rats were anesthetized with sodium pentobarbital (30 mg/kg, i.p.) at day 4 after treatment with PSLs. The peripheral blood (PB) was collected from the inferior vena cava of rats, and supplemented with novo-heparin (5 units/ml, Mochida Pharmaceutical, Tokyo, Japan). PB was carefully layered onto HISTOPAQUE 1083 (Sigma, MO, USA) and then centrifuged at 400 g for 30 min at 24°C. The mononuclear cell layer was taken out carefully after transferring the opaque interface, then centrifuged at 250 g for 10 min with PBS. Suspended cells in DMEM were incubated at 37°C in a 5% CO2 atmosphere for 3 h. The purity of monocytes was >98% as determined by the immunostaining of ED1 after removal of non-adherent cells. The adherent monocytes were cultured for another 24 h for further assays.
Real-Time Quantitative RT–PCR Analysis
Ankle joint tissues were obtained after the skin was removed. The total RNA from ankle join tissue and PB monocytes was extracted with the RNeasy Lipid Tissue Midi kit (QIAGEN, Germany), According to the manufacturer's instructions. A total of 400 ng of extracted total RNA was reverse transcribed to cDNA using the PrimeScript RT reagent kit (Takara, Shiga, Japan). The thermal cycling was holding at 95°C for 10 min, followed by 30–45 cycles of 95°C for 15 s and 60°C for 30 s. After the amplifying PCR reaction, the melting curve analysis was performed from 55 to 95°C (0.5°C/s). Real-time PCR was performed using the SYBR Premix Ex Taq (Takara) kit with a real-time PCR thermocycler (Rotor-Gene 3000A, Corbett Research, Australia). β-Actin was used as an internal control in each procedure. Primer sequences were as follows: RANKL: 5′-TCGGGTTCCCATAAAGTCAG-3′ and 5′-CTGAAGCAAATGTTGGCGTA-3′; RANK: 5′-ACCTGTCTTCTAAATGCACTC-3′ and 5′-CTTGCCTGCATCACAGA CTT-3′; osteoprotegerin (OPG): 5′-ACACACCAACTGCAGCTCAC-3′ and 5′-TGTCCACCAGAACACTCAGC-3′; IL-10: 5′-GAAGCTGAAGACCCTCTGGATACA-3′ and 5′-CCTTTGTCTTGGAGCTTATTAAAATCA-3′; IL-1β: 5′-CCTGTGGCCTTGGGCCTCAA-3′ and 5′-GGTGCTGATGTACCAGTTGGG-3′; IL-17: 5′-TTCTCCAGAACGTGAAGGTC-3′ and 5′-GGACAATAGAGGAAACGCAG-3′. The standard curve method was used for quantifying the levels of gene expression. The concentration ratio (target gene/β-actin) of AA rat samples treated with or without PSLs was compared with the sham samples. All real-time RT–PCR experiments were repeated three times and the results are represented as the mean of the ratio±s.d.
μCT Analysis
The ankle joints of sham, AA rats and the AA rats at day 18 after the treatment in each group (n=6) were examined using ScanXmate-E090S40 in vivo (Comscantecno, Kanazawa, Japan). Briefly, rat paws were scanned, the tibia, talus and calcaneus in the ankle joints were examined by three-dimensional surface rendering with a common threshold, and were optimized using histomorphometric techniques (GEMS MicroView). As the trabecular bone loss within the tibia and talus in the AA rats was too severe to quantify, calcaneuses were used for the bones qualitative analyses. The parameters of trabecular bone in each group were analyzed using TRI/3D-BON software program (Ratoc System Engineering, Tokyo, Japan).
Preparation of Peritoneal Macrophages
Rat peritoneal macrophages were collected with PBS 4 days after the injection of TGC (4.05 mg/kg, i.p.). The collected cells were washed twice with DMEM containing 4500 mg glucose/l, L-glutamine, NaHCO3 and pyridoxine (Sigma, D5796) and plated on plastic tissue culture dishes (2 × 106 cells). After incubation at 37°C in a 5% CO2 atmosphere for 3 h, the non-adherent cells were removed by washing the dishes three times with DMEM medium. The cell pattern was determined by cell morphology analysis with a light microscope. The total cells (95% macrophages) were incubated at 37°C/5% CO2.
Western Blotting
The macrophages were stimulated by lipopolysaccharide (LPS) (10 ng/ml) and treated with PSLs (100 μM) at the same time. The PSL-treated or untreated macrophage extracts were prepared with cell lysis buffer as described previously.13 A western blot analysis was performed with the following antibodies: rabbit polyclonal anti-p38 mitogen-activated protein kinase (p38 MAPK, 1:500 dilution), anti-p-p38 MAPK (1:500 dilution), anti-ERK (ERK1/2, 1:500 dilution) or anti-p-ERK1/2 (1:500 dilution). Bands were detected by the enhanced chemiluminescence detection system (ECL kit, Amersham Bioscience, Uppsala, Sweden) and quantified with an image analyzer FLA3000 (Fuji Photo Film, Tokyo, Japan). Values of p-p38 and p-ERK were normalized to total p38 and ERK values.
ELISA
The macrophages were activated by LPS (10 ng/ml) for 1 h, then treated with PSLs (100 μM) for another 3 h. Supernatants of the cultured cells were collected, and the release of IL-10 and IL-1β were measured using IL-10 and IL-1β ELISA kits (R&D, Minneapolis, MN, USA). The assay was performed according to the manufacturer's directions. The absorbence at 450 nm was performed using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA).
Immunohistochemistry and Histological Analysis
The sections of ankle joints from sham, AA and the PSL-treated AA rats were blocked with 10% normal donkey serum, and were incubated with goat anti-IL-10, anti-IL-1β, anti-IL-17, mouse anti-ED1 and anti-W3/25 antibodies for 18 h at 4°C. After incubating with a biotinylated anti-goat or anti-rabbit or anti-mouse IgG (1:200, Jackson Immunoresearch Laboratory, West Grove, PA, USA) for 2 h at 24°C, the sections were incubated with peroxidase-conjugated streptavidin (1:300, Dako, Japan, Kyoto, Japan) for 30 min at 24°C. The peroxidase was developed using 3, 3′-diaminobenzidine (DAB substrate kit, Vector Laboratory, Burlingame, CA, USA), and Mayer's hematoxylin as a counterstain. The normal goat, mouse or rabbit antibodies, which were substituted for primary antibodies, were used as negative controls. The immunoreactive cells were counted under a light microscope using the × 40 objective and × 10 ocular lenses in three areas per section. Histological analyses were performed using hematoxylin–eosin (H-E) and tartrate-resistant acid phosphatase (TRAP) staining (TRAP kit, Sigma-Aldrich).
Immunofluorescent Staining
To detect the cellular source of the cytokines in the ankle joints tissues, the sections were treated with 10% donkey serum for 1 h at 24°C, and then were incubated with each primary antibody for 18 h at 4°C. The primary antibodies were goat polyclonal anti-IL-10, anti-IL-1β, rabbit polyclonal anti-p-p38, anti-p-ERK and anti-p-IκBα mixed with mouse anti-ED1 or anti-W3/25 monoclonal antibodies. After washing with PBS, the sections were incubated with a mixture of FITC-conjugated and rhodamine-conjugated secondary antibodies for 1 h at 24°C. After washing, the sections were mounted in the anti-fading medium Vectashield (Vector Laboratory). The phagocytosis of PSLs by monocytes/macrophages in vivo and in vitro was also examined. In vivo, after 6 h of systemic treatment with NBD-labeled PSLs (5 mg/kg) or NBD-labeled PCLs (5 mg/kg) in AA rats, the animals were intracardially perfused with 2% PFA, and the ankle joints were removed and immersed in the same fixative for 12 h at 4°C. After immersing in 30% sucrose in PBS for 2 days, samples were embedded in an optimal cutting temperature compound (Sakura Finetechnical). Serial coronal frozen sections (10 μm) were prepared for immunohistochemistry. In vitro, NBD-labeled PSLs (100 μM) or NBD-labeled PCLs (100 μM) were applied to cultured peritoneal macrophages and PB monocytes on chamber slides (Nalge Nunc International, Rochester, NY, USA), and the cells were collected after 4 h. The cells were fixed with 2% PFA for 30 min at 24°C. After incubation with 10% donkey serum, the samples were incubated with mouse anti-ED1 IgG (1:200) for 18 h at 4°C. After washing with PBS, the slides were incubated with rhodamine-conjugated donkey anti-mouse IgG for 30 min at 24°C. The slides were mounted with Vectashield (Vector Laboratory). The samples were examined with a confocal laser scanning microscope, LSM510MET (Carl Zeiss, Jena, Germany).
Statistical Analysis
All values are expressed as the mean±s.d. Significant differences between groups were determined with Student's t-test. Differences with P-values of <0.05 were considered to be statistically significant.
RESULTS
The Effects of PSLs on the Paw Swelling, Bone Destruction and Expression of RANKL/RANK in the Ankle Joints of AA Rats
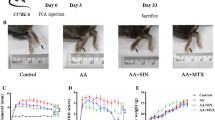
The paw volume increase of AA rats occurs in two stages: an acute inflammation stage from 10 to 21 days after CFA injection, and the chronic bone/joint destruction stage thereafter (Figure 1a). The paw volume of AA rats did not return to the control levels (Figure 1a and c). The CFA-induced clinical symptoms were similar to those in our previous studies.27, 28, 29 To focus on the effects of PSLs on AA-induced inflammatory bone loss, systemic treatment with PSLs was begun 10 days after CFA injection, when the inflammation was established without bone destruction in the ankle joint. Treatment with PSLs for 18 days significantly reduced the paw volume and resulted in a marked increase in the synovial thickness of ankle joints in AA rats during the chronic stage, without any significant effect during the acute stage (Figure 1a–c; horizontal bars). According to the three-dimensional surface rendering μCT analysis, the bone in the ankle joint including the tibia, talus and calcaneus in AA rats was markedly destructed in comparison with the sham rats (Figure 1d). The analysis of the parameters of trabecular bone was conducted in the calcaneus, because the trabecular bone losses within the tibia and talus in AA rats were too severe to quantify at 28 days after CFA injection. The mean trabecular bone volume per unit of metaphysis (BV/TV) was significantly decreased in AA rats to a similar degree as that observed in our previous experiments.17 Furthermore, numerous TRAP-positive osteoclasts were found in the bone cavity of the distal epiphysis of the tibia of AA rats (Figure 1e). Treatment with PSLs significantly promoted the recovery of the AA-induced paw volume increase (Figure 1a and b), the increase in the synovial thickness of ankle joints (Figure 1c; horizontal bars), bone destruction (Figure 1d) and the increased number of TRAP-positive osteoclasts (Figure 1e). However, PCLs had no effect on the paw volume of AA rats at any stage (Figure 1a).

Effects of systemic treatment with PSLs on paw swelling, trabecular bone loss and expression of RANKL/RANK in the ankle joints of AA rats. (a) The time course of the mean paw volume of AA rats after CFA injection. Each symbol and bar represents the mean±s.d. from six rats at each group. Sham: sham rats, AA: AA rats, AA+PSL: PSL-treated AA rats, AA+ PCL: PC liposome-treated AA rats. The asterisks indicate significant differences between AA and AA+PSL (*P<0.05, **P<0.01, Student's t-test). (b) Photographs showing the macroscopic appearances of ankle joints, (c) H-E staining of the synovium of ankle joints in AA rats. The horizontal bars indicate the thickness of the synovium. (d) Photographs showing the three-dimensional surface of bones in the ankle joints, including the tibia (Tb), talus (Tal) and calcaneus (Cal) according to the μCT analyses. (e) TRAP staining of the bone cavity of the epiphysis of the tibia, it is noted that there were numerous TRAP-positive mature osteoclasts in the AA rats (arrow heads). The mean mRNA levels of RANKL (f), RANK (g) and OPG (h) in the ankle joints in AA. Each column and bar represents the mean±s.d. from three rats at each group. The asterisks indicate significant differences between these values (*P<0.05, **P<0.01). Scale bar=50 μm.
The expression of RANKL, RANK and OPG was then examined, because these molecules have essential roles in trabecular bone loss in AA rats. The mean mRNA levels of RANKL and RANK were significantly increased in AA rats in comparison to sham rats, and treatment with PSLs for 18 days significantly decreased the AA-induced increased mean mRNA levels of both RANKL and RANK in the ankle joints of AA rats (Figure 1f and g). The mean mRNA level of OPG was unchanged after CFA injection, and PSLs had no effect on the expression of OPG of AA rats (Figure 1h).
Effects of PSLs on the Pro- and Anti-inflammatory Cytokine Production by the Infiltrated Inflammatory Cells in the Ankle Joints of AA Rats
At 6 h after intramuscular treatment, large granular aggregates of PSLs (white) were encircled by lysosomal membranes stained with the anti-ED1 antibody (gray) in the infiltrated macrophages in the synovium of ankle joints (Figure 2a). Interestingly, treatment with PSLs significantly reduced the total numbers of either ED1+ macrophages or W3/25+ helper T cells, which are the major infiltrated cells that are involved in the pathological process of RA in AA rats, in the synovium of ankle joints of AA rats at the chronic stage, without affecting their infiltration at the acute stage (Figure 2b).

Effects of systemic treatment with PSLs on the infiltration macrophages and helper T cells and the expression of IL-10, IL-1β and IL-17 in the ankle joints of AA rats. (a) Immunofluorescent CLSM images showing NBD-PSLs (white) are phagocytozed by ED1+ macrophages (gray) in the synovium of AA. (Scale bar=10 μm). (b) The mean number of ED1+ macrophages and W3/25+ helper T cells in the synovium of ankle joints of Sham, AA and AA+PSL. Each column and bar represents the mean±s.d. from six rats in each group. The asterisks indicate significant differences between the values (*P<0.05, **P<0.01, Student's t-test). (c–e) The mean mRNA levels of IL-10 (c), IL-1β (d) and IL-17(e) in the ankle joints of AA rats after systemic treatment with PSLs for 4 and 11 days. Each column and bar represents the mean±s.d. from six rats in each group. Asterisks indicate significant differences between the sham and AA animals (*P<0.05, **P<0.01, ***P<0.001, Student's t-test), and the swords indicate significant differences between AA and AA+PSL (†P<0.05, ††P<0.01, Student's t-test).
The failure of PSLs to inhibit the infiltration of immune cells into the ankle joints of AA rats at the acute stage led us to examine the effects of PSLs on the phenotypic change of infiltrated macrophages and helper T cells. In comparison with sham rats, the mean mRNA expression levels of IL-10 and IL-1β significantly increased in AA rats from day 14 to 21 after CFA injection (Figure 2c and d). However, IL-17, a major cytokine secreted by helper T cells, was also induced in the ankle joints of AA rats at 21 days after CFA injection (Figure 2e). Systemic treatment with PSLs was begun from day 10 after CFA injection. It was noted that treatment with PSLs for 4 and 11 days significantly increased the mean mRNA level of IL-10 (Figure 2c). On the other hand, PSLs significantly decreased the mean mRNA levels of IL-1β and IL-17 in the ankle joints of AA rats over same periods (Figure 2d and e).
According to the immunochemical analyses, the localization of the cytokines was determined in the synovium of ankle joints at the acute stage of AA rats (Figure 3). The mean number of IL-10-positive cells significantly increased 11 days after treatment with PSLs (Figure 3a, b and g). However, the mean numbers of IL-1β-positive cells and IL-17-positive cells in AA rats were significantly decreased (Figure 3c–f, h and i). No immunoreactivity was observed when normal rabbit IgG, normal goat IgG or normal mouse IgG was substituted for the primary antibodies as negative controls (data not shown). Furthermore, immunoreactivity for both IL-1β and IL-10 was mainly found in ED1+ macrophages (84 and 62%, respectively) and these cytokines were only partially expressed in W3/25+ helper T cells (8 and 18%, respectively) in the synovium of ankle joints at the acute stage of AA rats. We observed the immunoreactivity for IL-1β and IL-10 in ED1+ macrophages (43 and 86%, respectively) after treatment with PSLs (Figure 3l).

Effects of systemic treatment with PSLs on the immunoreactivity and localization of IL-10 and IL-1β in the ankle joints of AA rats at 21 days after CFA injection. (a–f) Immunoreactivities of IL-10 (a, b), IL-1β (c, d) and IL-17 (e, f) in the synovium of ankle joints of AA rats (AA) and PSL-treated AA rats (AA+PSL). (g, h, i) The mean number of positive cells for IL-10 (g), IL-1β (h) and IL-17 (i) in the synovium of ankle joints of AA and AA+PSL. Each column and bar represents the mean±s.d. from six rats in each group. Asterisks indicate significant differences between AA and AA+PSL (**P<0.01). Scale bar=20 μm (a–f). (j–k) Identification of IL-1β- and IL-10-expressing cells in the ankle joint in AA. Immunofluorescent CLSM images for IL-1β (red) and ED1 (green) (j); IL-10 (red) and ED1 (green) (k). (l) The mean ratios of IL-1β and IL-10 in ED1+ macrophages infiltrated in the synovium of ankle joints of AA and AA+PSL. Scale bar=10 μm (j, k). Each column and bar represents the mean±s.d. from six rats in each group. Asterisks indicate significant differences between AA and AA+PSL (*P<0.01, ***P<0.001).
These observations strongly suggested that approximately half of infiltrated macrophages underwent phenotypic change from IL-1β-producing pro-inflammatory cells to IL-10-producing anti-inflammatory cells after the phagocytosis of PSLs.
PSLs Triggered LPS-Activated Macrophages to Anti-inflammatory Phenotype through Regulating MAPKs and NF-κB Activity
To clarify the mechanisms underlying PSL-induced phenotypic change of macrophages infiltrated in the ankle joints of AA rats, the effect of PSLs on the secretion of IL-10 and IL-1β from LPS-stimulated macrophages was examined, because LPS is a key component of CFA that activates macrophages in AA rats.31 Although the mean levels of IL-10 and IL-1β in the culture medium were relatively low in the none treated control macrophages, the mean levels were significantly increased after treatment with LPS (Figure 4a and b). PSLs significantly increased the IL-10 levels, but significantly decreased the IL-1β levels in the culture medium of LPS-activated macrophages (Figure 4a and b). These in vitro results were consistent with the observations from AA rats.

PSLs trigger LPS-stimulated macrophages to anti-inflammatory phenotype through the down-regulation of p-38 and NF-κB activity. (a, b) The mean levels of IL-10 (a) and IL-1β (b) in the culture medium of LPS-stimulated macrophages. None: non-treated cells, LPS: LPS-stimulated cells, LPS+PSL: PSL-treated LPS-stimulated cells. Each column represents the mean of three separate experiments. Asterisks indicate significant differences between the values (*P<0.05 **P<0.01). (c) A western blot analysis of p38 MAPK and ERK in LPS-stimulated macrophages after treatment with PSLs. (d). The mean relative ratio of p38 MAPK and ERK activation. Each column represents the mean of three separate experiments. LPS: LPS stimulation, LPS+PSL: LPS stimulation on PSL-treated cells. Asterisks indicate significant differences after LPS stimulation (*P<0.05 **P<0.01). Swords indicate significant differences between LPS and LPS+PSL (†P<0.05). (e) Immunofluorescent CLSM images indicating the nuclear translocation of NF-κB (green) in macrophages with DAPI-stained nuclei (blue). None: non-treated cells, LPS: LPS-stimulated cells, LPS+PSL: PSL-treated LPS-stimulated cells. Scale bar=10 μm.
The effects of PSLs on the activation of p38 MAPK, ERK and NF-κB, which have key regulatory roles in the cytokine production, were next examined. Upon LPS stimulation, p38 MAPK was activated rapidly after 15–30 min. In contrast, ERK was activated slowly from 30 min and lasted up to 60 min (Figure 4c). Furthermore, the numerous nuclear translocation of NF-κB was detected at 30 min after treatment with LPS. After treatment with PSLs, the mean level of p-p38 MAPK significantly decreased from 15 to 30 min (Figure 4c and d). However, PSLs significantly increased the mean level of p-ERK from 30 to 60 min (Figure 4c and d). Moreover, PSLs markedly inhibited the LPS-induced nuclear translocation of NF-κB (Figure 4e). These observations indicated that PSLs inhibited the LPS-induced activation of p38 MAPK and NF-κB, but enhanced LPS-induced activation of ERK, thus resulting in the shift of the LPS-stimulated macrophages from IL-1β-producing pro-inflammatory cells into IL-10-producing anti-inflammatory cells.
The effects of PSLs on the production of both IL-10 and IL-1β by the PB monocytes of AA rats were further examined, because the PB monocytes are the sources of the macrophage infiltrating the synovium of ankle joints of AA rats. PSLs were phagocytosed by the ED1+ PB monocytes of AA rats (Figure 5a). The mean mRNA levels of both IL-10 and IL-1β in the PB monocytes of AA rats were markedly increased in comparison with that in sham rats (Figure 5b and c). Treatment with PSLs significantly increased the mean expression level of IL-10, but significantly decreased that of IL-1β in the PB monocytes of AA rats (Figure 5b and c).

PSLs induce the phenotypic change of infiltrated macrophages in AA rats from the pro-inflammatory to the anti-inflammatory phenotype through the inhibition of p-38 and NF-κB activity. (a) Immunofluorescent CLSM images showing NBD-PSL (green) are phagocytozed by the PB ED1+ monocytes (red) of AA rats (Scale bar=5 μm). (b, c) The mean mRNA levels of IL-10 (b) and IL-1β (c) in the PB ED1+ monocytes of AA rats after systemic treatment with PSLs for 4 days. Each column and bar represents the mean±s.d. from six rats in each group. Asterisks indicate significant differences between the AA and AA+PSL (*P<0.05, **P<0.01, Student's t-test). (d) Immunofluorescent CLSM images for p-p38 (red) and ED1 (green); p-ERK (red) and ED1 (green); p-IκBα red) and ED1 (green) (Scale bar=20 μm). (e–g) The mean percentage of p-p38-, p-ERK- or p-IκBα-positive ED1+ macrophages and W3/25+ helper T cells inflated in the synovium of ankle joints of AA and AA+PSL rats. Each column and bar represents the mean±s.d. from six rats in each group. Asterisks indicate significant differences between AA and AA+PSL (*P<0.05, **P<0.01, Student's t-test).
Finally, the effects of PSLs on the activation of p38 MAPK, ERK and IκBα in the infiltrated macrophages and T cells in the synovium were examined. In AA rats, >70% of the ED1+ macrophages, as well as W3/25+ helper T cells, expressed p-p38 (Figure 5d and e), whereas only 30% of ED1+ and W3/25+ cells expressed p-ERK (Figure 5d and f). Furthermore, approximately 60% of ED1+ as well as W3/25+ cells expressed p-IκBα (Figure 5d and g). After treatment with PSLs, the mean percentage of p-p38-positive ED1+ cells and p-IκBα-positive ED1+ cells were significantly decreased (Figure 5d, e and g). On the other hand, the mean percentage of p-ERK-positive ED1+ cells was significantly increased (Figure 5d and f). In contrast, the mean percentage of neither p-p38, p-ERK nor p-IκBα-positive W3/25+ cells was significantly changed by treatment (Figure 5d–g). These in vitro and in vivo results indicated that PSLs directly induced the cell-specific phenotypic changes of the macrophages that had infiltrated the ankle joints of AA rats, changing it from an inflammatory phenotype to an anti-inflammatory phenotype.
DISCUSSION
The major finding of the current study was that PSLs inhibited the AA-induced trabecular bone loss partially through the phenotypic change of infiltrated macrophages from IL-1β-producing pro-inflammatory cells to IL-10-producing anti-inflammatory cells. To the best of our knowledge, this is the first report to address the effect of PSLs on the phenotypic change of macrophages that had infiltrated into the ankle joints of AA rats during inflammatory bone loss.
Pro-inflammatory cytokines produced by macrophages and helper T cells, the major infiltrated cells in the synovium of joints, have pivotal roles in the pathological process of RA.30, 31 AA, an animal model of RA, is widely used for studies of both inflammation and bone loss,18, 32 because AA rats show unique sequential pathological processes. In the acute stage, numerous macrophages and helper T cells infiltrate into the synovium, but bone loss is not detected. In the chronic stage, trabecular bone loss occurs, and the number of infiltrated macrophages and helper T cells declines.26, 28 In this study, the expression levels of IL-1β and IL-17 were markedly elevated in the ankle joints during the acute stage in AA rats. It was also noted that the production of IL-1β increased prior to that of IL-17. This was consistent with the fact that IL-1β is essential for promoting Th17 cell differentiation and IL-17 production by the differentiated Th17 cells.33 The expression level of IL-10 also significantly increased during the same period. Treatment with PSLs significantly decreased the IL-1β and IL-17 levels, and also significantly increased the IL-10 levels. It was considered that IL-10 secreted from macrophages after phagocytosis of PSLs can inhibit IL-1β production and the differentiation of Th17 cells, thus resulting in a decrease in the IL-17 levels.34 This PSL-induced phenotypic change of infiltrated macrophages also contributed to the inhibition of AA-induced trabecular bone loss through the inhibition of RANKL/RANK expression, which have essential roles in osteoclastogenesis18, 35, 36 and osteoclastic bone loss in RA as well as in AA.37 Treatment with PSLs significantly decreased the elevated levels of RANKL and RANK in AA rats, which occurred in parallel with a significant reduction of trabecular bone loss in AA rats. It is considerable that there are at least three possible mechanisms underlying PSL-induced down-regulation of RANKL/RANK in AA rats. First, PSLs decreased the expression levels of IL-1β and IL-17, which can promote the expression of RANKL.38, 39 Second, PSL-induced IL-10, which strongly inhibited the expression of RANKL/RANK.23, 37 Third, PSLs are phagocytosed by the osteoclast precursor to directly decrease the expression of RANK by osteoclast precursor and the production of RANKL in cultured rat bone marrow cells.17 Therefore, it is considered that the inhibitory effect of PSLs on the trabecular bone loss in AA rats completely depends on the inhibition of the RANKL/RANK pathway. However, a possibility that decreased RANKL/RANK expression may simply be due to the decrease in levels of inflammation cannot be totally ruled out.
LPS, one of constituents of CFA, can activate MAPKs and NF-κB.40 The activity of MAPKs and NF-κB are involved in the regulation of cytokine production.41, 42, 43 According to our experimental design, a low concentration of LPS induced the activation of p38 MAPK, ERK and NF-κB in macrophages resulted in the release of IL-1β and IL-10. It was noted that PSLs rapidly reduced LPS-induced activation of p38 MAPK, but enhanced LPS-induced activation of ERK. This is the first study to examine the effects of PSLs on LPS-induced MAPK activation. These observations are consistent with previous observations that PSLs induce ERK, but not p38 MAPK activity in primary cultured rat microglia13 and macrophages.44 Therefore, it is reasonable to consider that lysosomal proteolytic functions are involved in LPS-induced MAPKs activity following phagocytosis of PSLs. The inhibitory effects of PSLs on IL-1β secretion from LPS-activated macrophages may depend on the reduction of p38 MAPK activity, because p38 MAPK activity has crucial roles in the regulation of IL-1β biosynthesis45 and p38 MAPK inhibitor blocks IL-1β production.46 PSL-induced inhibition of NF-κB activity may be involved in the inhibition of IL-1β, because NF-κB activity is required for pro-inflammatory cytokine production, including IL-1β.43 However, PSL-induced increase in IL-10 by LPS-stimulated macrophages may depend on ERK activity, because ERK activity leads to IL-10 production.47, 48 Furthermore, PSLs inhibited the rapid activation of p38 MAPK and may result in the subsequent slow enhancement of ERK activity in LPS-stimulated macrophages, because p38 MAPK and ERK cross-regulate each other such as the inhibition of one enhances the activation of the other.47, 49 Moreover, PSLs regulated the expression levels of IL-10 and IL-1β in the PB monocytes, the source of macrophage infiltrated in the ankle joints, and regulated the phosphorylation of p38, ERK and IκBα in the infiltrated macrophages in the synovium of the ankle joints of AA rats. Therefore, it is considered that systemic treatment with PSLs might have effect on both the circulating and infiltrated monocytes/macrophages. These in vitro and in vivo observations strongly suggest that PSL-induced inhibition of p38 MAPK and the subsequent enhancement of ERK is the most likely underlying mechanism for phenotypic change of the infiltrated macrophages.
In conclusion, this study shows that systemic treatment with PSLs inhibited AA-induced trabecular bone loss by provoking the phenotypic change of macrophages from IL-1β-producing to IL-10-producing cells, in the ankle joint of AA rats. This phenotypic change of infiltrated macrophages is caused by the PSL-induced inhibition of p38 MAPK activity and the subsequent enhancement of ERK activity. As PS is a competent of the cell membrane, PSLs may be potentially useful for achieving pharmacological intervention against inflammatory bone loss in RA and periodontitis without any apparent deleterious side effects.
References
Fadok VA, Bratton DL, Konowal A, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J Clin Invest 1998;101:890–898.
Huynh ML, Fadok VA, Henson PM . Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J Clin Invest 2002;109:41–50.
Serhan CN, Savill J . Resolution of inflammation: the beginning programs the end. Nat Immunol 2005;6:1191–1197.
Serhan CN, Brain SD, Buckley CD, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J 2007;21:325–332.
Verhoven B, Krahling S, Schlegel RA, et al. Regulation of phosphatidylserine exposure and phagocytosis of apoptotic T lymphocytes. Cell Death Differ 1999;6:262–270.
Schlegel RA, Callahan MK, Williamson P . The central role of phosphatidylserine in the phagocytosis of apoptotic thymocytes. Ann N Y Acad Sci 2000;926:217–225.
Fadok VA, Bratton DL, Rose DM, et al. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000;405:85–90.
Hoffmann PR, deCathelineau AM, Ogden CA, et al. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol 2001;155:649–659.
Byrne A, Reen DJ . Lipopolysaccharide induces rapid production of IL-10 by monocytes in the presence of apoptotic neutrophils. J Immunol 2002;168:1968–1977.
Steinman RM, Turley S, Mellman I, et al. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med 2000;191:411–416.
Chen X, Doffek K, Sugg SL, et al. Phosphatidylserine regulates the maturation of human dendritic cells. J Immunol 2004;173:2985–2994.
Otsuka M, Tsuchiya S, Aramaki Y . Invilvement of EK, A MAP kinase, in the production of TGF-β by macrophages treated with liposomes composed of phosphatidylserine. Biochem Biophys Res Commun 2004;324:1400–1405.
Zhang J, Fujii S, Wu Z, et al. Involvement of COX-1 and up-regulated prostaglandin E synthases in phosphatidylserine liposome-induced prostaglandin E2 production by microglia. J Neuroimmunol 2006;172:112–120.
Shi D, Fu M, Fan P, et al. Artificial phosphatidylserine liposome mimics apoptotic cells in inhibiting maturation and immunostimulatory function of murine myeloid dendritic cells in response to 1-chloro-2, 4-dinitrobenze in vitro. Arch Dermatol Res 2007;299:327–336.
Hoffmann PR, Kench JA, Vondracek A, et al. Interaction between phosphatidylserine and the phosphatidylserine receptor inhibits immune responses in vivo. J Immunol 2005;174:1393–1404.
Ramos GC, Fernandes D, Charão CT, et al. Apoptotic mimicry: phosphatidylserine liposomes reduce inflammation through activation of peroxisome proliferator-activated receptors (PPARs) in vivo. Br J Pharmacol 2007;151:844–850.
Wu Z, Ma HM, Kukita T, et al. Phosphatidylserine-containing liposomes inhibit the differentiation of osteoclasts and trabecular bone loss. J Immnunol 2010;184:3191–3201.
Kong YY, Feige U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 1999;402:304–309.
Ji H, Pettit A, Ohmura K, et al. Critical roles for interleukin 1 and tumor necrosis factor-α in antibody-induced arthritis. J Ex Med 2002;196:77–85.
Karouzakis E, Neidhart M, Gay RE, et al. Molecular and cellular basis of rheumatoid joint destruction. Immunol Lett 2006;106:8–13.
Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synoviuml fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 1999;103:1345–1352.
Chabaud M, Miossec P . The combination of tumor necrosis factor alpha blockade with interleukin-1 and interleukin-17 blockade is more effective for controlling synoviuml inflammation and bone resorption in an ex vivo model. Arthritis Rheum 2001;44:1293–1303.
van de Loo FA, van den Berg WB . Gene therapy for rheumatoid arthritis. Lessons from animal models, including studies on interleukin-4, interleukin-10, and interleukin-1 receptor antagonist as potential disease modulators. Rheum Dis Clin North Am 2002;28:127–149.
Zhang X, Teng YT . Interleukin-10 inhibits gram-negative-microbe-specific human receptor activator of NF-κB ligand-positive CD4+ Th1-cell-associated alveolar bone loss in vivo. Infect Immun 2006;74:4927–4931.
Terrier F, Hricak H, Revel D, et al. Magnetic resonance imaging and spectroscopy of the periarticular Inflammatory soft-tissue changes in experimental arthritis of the rat. Invest Radiol 1985;20:813–823.
Wu Z, Nagata K, Iijima T . Immunohistochemical study of NGF and its receptors in the synoviuml membrane of the ankle joint of adjuvant-induced arthritic rats. Histochem Cell Biol 2000;14:453–459.
Toh K, Kukita T, Wu Z, et al. Possible involvement of MIP-1α in the recruitment of osteoclast progenitors to the distal tibia in rats with adjuvant-induced arthritis. Lab Invest 2004;84:1092–1102.
Wu Z, Nagata K, Iijima T . Involvement of sensory nerves and immune cells in osteophyte formation in the ankle joint of adjuvant arthritic rats. Histochem Cell Biol 2002;118:213–220.
Wu Z, Zhang J, Nakanishi H . Leptomeningeal cells activate microglia and astrocytes to induce IL-10 production by releasing pro-inflammatory cytokines during systemic inflammation. J Neuroimmunol 2005;167:90–98.
Klareskog L, Ronnelid J, Holm G . Immunopathogenesis and immunotherapy in rheumatoid arthritis: an area in transition. J Inter Med 1995;238:191–206.
Muller-Ladner U, Pap T, Gay RE, et al. Mechanisms of disease: the molecular and cellular basis of joint destruction in rheumatoid arthritis. Nat Clin Pract Rheumatol 2005;1:102–110.
Asagiri M, Hirai T, Kunigami T, et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science 2008;319:624–627.
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, et al. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol 2007;8:942–949.
Chang H, Hanawa H, Yoshida T, et al. Alteration of IL-17 related protein expressions in experimental autoimmune myocarditis and inhibition of IL-17 by IL-10-Ig fusion gene transfer. Circ J 2008;72:813–819.
Dougall WC, Glaccum M, Charrier K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev 1999;13:2412–2424.
Li J, Sarosi I, Yan XQ, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci 2000;97:1566–1571.
Gravallese EM, Manning C, Tsay A, et al. Synoviuml tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum 2000;43:250–258.
Suda T, Takahashi N, Udagawa N, et al. Modulation of osteoclast differentiation and function by the new member of the tumor necrosis factor receptor and ligan families. Endocrine Rev 1999;20:345–357.
Ju JH, Cho ML, Moon YM, et al. IL-23 induces receptor activator of NF-κB ligand expression on CD4+ T cells and promotes osteoclastogenesis in an autoimmune arthritis model. J Immunol 2008;181:1507–1518.
Sweet MJ, Hume DA . Endotoxin signal transduction in macrophages. J Leukoc Biol 1996;60:8–26.
Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994;372:739–746.
Feng GJ, Goodridge HS, Harnett MM, et al. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lipopolysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. J Immunol 1999;163:6403–6412.
Simmonds RE, Foxwell BM . Signalling, inflammation and arthritis: NF-κB and its relevance to arthritis and inflammation. Rheumatology 2008;47:584–590.
Aramaki Y, Matsuno R, Tsuchiya S . Involvement of p38 MAP kinase in the inhibitory effects of phosphatidylserine liposomes on nitric oxide production from macrophages stimulated with LPS. Biochem Biophys Res Commun 2001;280:982–987.
Baldassare JJ, Bi Y, Bellone CJ . The role of p38 mitogen-activated protein kinase in IL-1β transcription. J Immunol 1999;162:5367–5373.
Lee SJ, Lim KT . Phytoglycoprotein inhibits interleukin-1β and interleukin-6 via p38 mitogen-activated protein kinase in lipopolysaccharide-stimulated RAW 264.7 cells. Naunyn Schmiedebergs Arch Pharmacol 2008;377:45–54.
Mathur RK, Awasthi A, Wadhone P, et al. Reciprocal CD40 signals through p38 MAPK and ERK-1/2 induce counteracting immune responses. Nat Med 2004;10:540–544.
Lucas M, Zhang X, Prasanna V, et al. ERK activation following macrophage FcγR ligation leads to chromatin modifications at the IL-10 locus. J Immunol 2005;175:469–477.
Kim C, Sano Y, Todorova K, et al. The kinase p38α serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat Immunol 2008;9:1019–1027.
Acknowledgements
We thank the staff from Kaken pharmaceutical company for the helpful μCT technique. This work was supported by Grants-in-Aid for Scientific Research (No. 20592174 to Wu Z; No. 20390472 to Nakanishi H).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Local treatment with phosphatidylserine-containing liposomes (PSLs) ameliorate inflammatory bone loss induced by adjuvant-injection. Infiltrated macrophages undergo phenotypic change from IL-1β-producing to IL-10-producing cells after phagocytosis of PSLs. The likely underlying mechanism is that PSL has differential effects on the activities of p38 MAPK and ERK.
Rights and permissions
About this article
Cite this article
Ma, H., Wu, Z. & Nakanishi, H. Phosphatidylserine-containing liposomes suppress inflammatory bone loss by ameliorating the cytokine imbalance provoked by infiltrated macrophages. Lab Invest 91, 921–931 (2011). https://doi.org/10.1038/labinvest.2011.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2011.54
Keywords
This article is cited by
-
Increased n-6 Polyunsaturated Fatty Acids Indicate Pro- and Anti-Inflammatory Lipid Modifications in Synovial Membranes with Rheumatoid Arthritis
Inflammation (2023)
-
LILRB2-mediated TREM2 signaling inhibition suppresses microglia functions
Molecular Neurodegeneration (2022)
-
Enhancing effect of sodium butyrate on phosphatidylserine–liposome-induced macrophage polarization
Inflammation Research (2022)
-
Effect of pegylated phosphatidylserine-containing liposomes in experimental chronic arthritis
BMC Pharmacology and Toxicology (2015)
-
Myelin alters the inflammatory phenotype of macrophages by activating PPARs
Acta Neuropathologica Communications (2013)
