
Abstract
Hepatic fibrosis is the result of chronic liver injuries underlined by diverse etiologies. The massive accumulation of extracellular matrix (ECM) proteins during fibrogenesis leads to structural distortion and functional disruption of the liver. There is currently no effective standard treatment for liver fibrosis. We previously identified a class of imidazolium salts (IMSs) with anti-fibrotic properties in a cell-based screen. In this report, we investigated the anti-fibrotic efficacy and mechanisms of a small IMS, 1,3-diisopropylimidazolium tetrafluoroborate (DPIM), in a hepatic fibrosis model induced by bile duct ligation (BDL) in mice. The orally available DPIM was administered to BDL mice via drinking water at three concentrations (0.5, 0.75, and 1 g/l) for 4 weeks. We observed a significant reduction in inflammation and collagen deposition in the liver, which could be mediated by a reduction in the expression of monocyte chemoattractant protein-1 (MCP-1) and by an enhancement in the matrix metalloproteinase-mediated ECM remodeling. The current findings highlight the importance for simultaneously targeting multiple pathways to more effectively attenuate and resolve liver fibrosis and warrant further studies on this compound in additional models of hepatic fibrosis.
Similar content being viewed by others


Main
Hepatic fibrosis represents a major health burden worldwide. Hepatic fibrosis is an abnormal liver wound-healing response during persisting liver injuries triggered by wide varieties of stimuli, such as toxic damage, viral infection, metabolic disorders, and alcohol abuse. Among the many possible pathological pathways that may contribute to fibrogenesis,1 inflammation and extracellular matrix (ECM) accumulation have been recognized as two of the pivotal players. A linear correlation between the expression of monocyte chemoattractant protein-1 (MCP-1 or CCL2) and the infiltration of CD68+ immune cells (leukocytes, macrophages, and Kupffer cells) was observed in the fibrotic human liver tissues.2 Experimentally, the MCP-1 receptor, CC-chemokine receptor 2 (or CCR2), expressed on the surface of macrophages, Kupffer cells, and hepatic stellate cells (HSCs), was found to mediate fibrogenesis during liver injuries.3 In contrast, suppressing macrophage infiltration resulted in an inhibition of HSC activation, as triggered by liver injury, which in turn could ultimately improve the recovery outcome.4 Intriguingly, the inflammatory MCP-1 was found to induce the production of fibrogenic collagen in portal fibroblasts,5 which prominently contribute to the fibrogenesis in cholestatic injuries.6 In parallel, pathological changes in ECMs, as consequences of an alteration in the delicate balance between the matrix metalloproteinases (MMPs) and the tissue inhibitors of metalloproteinases (TIMPs), also significantly contribute to fibrogenesis, and to a great extent dictate the recovery outcomes.7 Based on such an analysis, therapeutic strategy aimed at restoring appropriate ECM compositions by regulating MMPs and TIMPs has also been attempted to attenuate and to resolve fibrosis.8, 9
Despite our extensive knowledge on the importance of inflammation and ECM remodeling in fibrogenesis and resolution, no effective drug therapy has been reported by simultaneously targeting these two key aspects with small molecules. In a recent study,10 we have identified several imidazolium salts (IMSs) that can reduce collagen production in a cell-based screening, and further shown that one of the IMSs, 1,3-bisbenzylimidazolium bromide (DBZIM), can reduce the collagen content in the liver of a mouse cholestasis model. In the same cell-based screen, we also uncovered a simpler IMS, 1,3-diisopropylimidazolium tetrafluoroborate (DPIM), which is devoid benzyl substituent, with an anti-fibrotic effect. The objective of the current study is to investigate whether DPIM possesses anti-fibrotic efficacy in the same mouse cholestasis model, and the possible anti-fibrotic mechanisms associated with DPIM.
MATERIALS AND METHODS
Compound
The IMS DPIM (empirical formula C9H17N2 BF4; MW 240. 05; Cat. No. 660019) was purchased from Sigma-Aldrich Chemicals (St Louis, USA), and its chemical structure was depicted in Figure 1a. It was dissolved in cell culture medium for in vitro work or in drinking water for animal experiment.

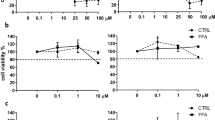
DPIM and treatment in HSC-T6 cells and mice. (a) Chemical structure of DPIM. (b) Total proteins isolated from HSC-T6 cells treated with three concentrations (50, 100, and 200 μM) of DPIM for 48 h were immunoblotted for Col1a1 and α-SMA. The transcript levels for α-SMA (c) and Col1a1 (d) were measured by real-time PCR. *P<0.05, **P<0.01.
Cell Culture
The HSC-T6 cells, kindly provided by Dr Scott Friedman of Mount Sinai School of Medicine (New York), were cultured in DMEM media supplemented with 10% fetal bovine serum (FBS), 100 IU/ml penicillin/100 μg/ml streptomycin. Cells at around 10% confluency were treated with different concentrations of DPIM compound for 2 days. Total RNA and protein were isolated for real-time PCR and western blotting analysis, respectively. All cell culture reagents were from Invitrogen (CA, USA). BrdU cell proliferation assay (Cat. No. 2759, Millipore, USA) was conducted according to the manufacturer's instructions. The HSC-T6 cells were treated with DPIM (0–200 μM) for 48 h, and BrdU was added into the culture 24 h before the assay.
Gene Expression Profiling
HSC-T6 cells initially seeded in T75 flasks in regular DMEM (with 10% FBS) were changed to a low serum (0.5%) DMEM at a confluence of 70–80% and adapted for an additional 12 h. Cells were then incubated with 50, 100, and 200 μM of DPIM for 48 h. Total RNA was extracted using a NucleoSpin RNA II kit (Macherey-Nagel, Germany) and quantified using the ND-100 spectrophotometer (Nanodrop Technologies, DE, USA). RNA quality was verified using the eGENE cartridge QIAxcel RNA Quality Control Kit (Qiagen). Reverse transcription and amplification to cRNA was performed using the Illumina TotalPrep RNA Amplification Kit (Ambion). cRNA was then hybridized in a shaking oven with the Rat Ref-12 chip (Illumina) for 20 h at 58 °C. The chip was washed and scanned using the BeadScan hardware (Illumina). The initial data processing was done using the BeadStudio V2.0 software to subtract the background noise and then to normalize the signal to average signal of the total chip's array. All data were subtracted from background intensities and were normalized using the cross-correlation method.11 For each gene, a fold-change value was calculated for compound treatments versus control. Differentially expressed genes (DEGs) were selected based on the criterion of fold change >2. The P-values of DEGs were obtained using one-tailed Student's t-test. Pathway analysis was carried out on the DEGs using ingenuity pathway analysis (Ingenuity Systems, CA, USA). Detailed results were displayed in Supplementary Figure 1.
Bile Duct Ligation and Compound Treatment in Mice
Adult (8–10 weeks) male mice of FVB/N background were used for the experiments. The bile duct ligation (BDL) surgery was performed as previously described.10 The BDL mice and sham-operated mice were assigned to five different treatment groups (Supplementary Figure 3), with 6–8 mice in each group. All mice (sham or BDL) had free access to drinking water with (0.5, 0.75, or 1 g/l) or without DPIM, as indicated in Supplementary Figure 3. On average, each mouse (BDL or sham) drank ∼3.5 ml of water daily, regardless of the absence or presence of the compound. All animals undergone BDL procedure were allowed to live until the fourth week, if the animals survived through the first 3 days after the surgery. At the end of the fourth week, all mice were killed and liver tissues were collected for sectioning or other biochemical assays. All the procedures performed on mice were approved by a local IACUC committee.
Liver Section, Sirius Red Staining, and Immunohistochemistry
Paraffin sectioning, Sirius red staining, and quantification were performed as previously described.10 To ensure sampling accuracy, at least six different areas from the left and middle lobes of each liver were included for imaging and quantification. We chose to use the Sirius Red stain to better visualize the assembled collagen fibers, since degraded collagen fragments is also reflected in the hydroxyproline assay.12 The H&E staining was performed to analyze liver architecture and the presence of inflammatory cells.
Frozen liver sections (16 μm) were prepared for immunostaining with anti-α-SMA (Cy3 labeled, 1:200 dilution, Sigma), anti-CD68 (1:100 dilution, Serotech, USA), anti-cytokeratin (1:200 dilution, Z6022, DAKO, Denmark), respectively. The secondary antibodies, anti-rat Alexa488 and anti-rabbit Alexa555 (Invitrogen), were used at a dilution of 1:200. Fluorescent images were captured using the LEICA RMB-DM epifluorescence microscope (LEICA, Germany).
Protein Extraction, SDS–PAGE, and Immunoblot
Total protein extraction from HSC-T6 or liver tissue was performed as previously described.10 Primary antibodies used for the western blots were α-SMA (1:1000 dilution, Sigma), α-Tubulin (1:2000 dilution, Santa Cruz Biotech, USA), TIMP-1 (1:500 dilution, sc-80363, Santa Cruz Biotech), MCP-1 (1:500 dilution, sc-28879, Santa Cruz Biotech).
Quantitative Real-Time PCR for Gene Expression Analysis
Total RNA was extracted from the HSC-T6 cells or liver tissues according to the manufacturer's protocol (RNeasy kit, Machery-Nagel, Germany). Total RNA (1.2 μg) was then reverse transcribed to cDNA by Superscript III reverse transcriptase (Invitrogen) with random hexamer in a total reaction volume of 20 μl. Real-time PCR reactions (in triplicates) were performed with the Fast Real-Time PCR System (Applied Biosystems, USA), using 1 μl of cDNA product in a reaction volume of 10 μl. The Taqman primers and probes (Applied Biosystems) for target genes, α-SMA, collagen 1A1, TGFβ1, TIMP-1, MMP-2, MMP-8, MMP-9, MMP-13, MCP-1, and the endogenous control β-actin were Mm01204962_GH, Mm00801666_g1, Mm00441724_m1, Mm00441818_m1, Mm00439509_m1, Mm00439506_m1, Mm00442991_m1, Mm01168713, Mm00441243_g1, respectively. The PCR conditions were 95 °C for 20 s and 40 cycles of amplification at 95 °C for 3 s and 60 °C for 30 s. The transcript levels were expressed as fold of changes compared with sham control unless stated otherwise.
Measurement of MMP Activities and Zymography
Total MMPs activities were measured using fluorogenic peptide substrate. Liver samples were homogenized in PBS buffer without adding exogenous proteinase inhibitors, and 50 μg of total proteins were applied to the peptide substrate (ES010, R&D, USA) in a black 96-well plate.13 After a 30-min incubation, the fluorescent intensity was measured with a microplate reader (Safire II, TECAN) with excitation at 320 nm and emission at 405 nm. The fluorescent activities were calculated as relative level compared with BDL liver samples.
MMP-2 and MMP-9 activities were measured by the gelatin zymography (Invitrogen). Fifty micrograms of total liver proteins were first subject to electrophoresis in a 10% SDS–PAGE gel containing 0.1% gelatin. The gel was then re-natured, developed, and stained with colloidal blue, according to the manufacturer's instructions (Invitrogen).
Measuring Glutathione (GSH) and the Ratio of GSH/Disulphide Dimer of Glutathione (GSSG)
Total cellular GSH and GSSG levels were determined as previously described.10
TUNEL Staining
Liver paraffin sections (in 5 μm thickness) were stained for apoptotic cells using a TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) Kit (TA4627, R&D system) according to the manufacturer's instructions. The slides were subsequently counterstained with DAPI and mounted with VECTASHIELD Fluorescent Mounting Media (H-1000, Vector Labs), before imaging on a Leica DM IRB inverted microscope.
Biochemical Analysis of Serum Parameters
At the end of experiment (the fourth week), blood samples were collected by retro-orbital bleeding, and the serum alanine aminotransferase (ALT), AST, ALP, bilirubin, GGT activities were measured on the Cobase c 111 analyzer (Roche Diagnostic, USA) according to the manufacturer's instructions.
Statistical Analysis
All images displayed are representative of 6–8 mice, unless stated otherwise. All quantitative data were expressed as the mean±s.d., and comparisons between pairs were analyzed with two-tailed Mann–Whitney U-test. P-values <0.05 were considered statistically significant.
RESULTS
Effects of DPIM in HSC-T6 Cells
We had previously showed that DPIM at 1 mM (IC50=3.1 mM) significantly reduced the expression of Col1α1 and α-SMA in the HSCs-T6 cells.10 Here, we confirmed that DPIM at much lower concentrations (50, 100, and 200 μM) for 48 h also dose dependently suppressed the expression of the fibrogenic Col1α1 gene, but not the α-SMA (Figures 1b–d) after 48 h of incubation. To survey global gene expression, HSC-T6 cells treated with the same conditions (50, 100, and 200 μM of DPIM for 48 h) were subject to gene profiling using the Illumina chips. The bioinformatic analysis indicated that a number of signaling pathways relevant inflammation and fibrosis in cultured HSCs were significantly affected by the DPIM treatment (Supplementary Figure 1). Further real-time RT–PCR experiments confirmed that the transcription of TIMP-1 and MCP-1 was suppressed in the HSC-T6 cells treated with DPIM (200 μM for 48 h), accompanied by a significant increase in MMP-9 (gelatinase) and MMP-13 (collagenase) (Supplementary Figure 2a). However, DPIM treatment (10, 100, 200 μM for 48 h) failed to significantly inhibit the proliferation of HSC-T6 cells (Supplementary Figure 2b), albeit a slight decrease in proliferation was achieved at 200 μM (P=0.08, compared with 0 μM). With this pathway information at hand, we next investigated the expression of several molecules known to have pivotal roles in inflammatory chemotaxis and ECM remodeling during liver injury and recovery in vivo using the dosing scheme outlined in Supplementary Figure 3.
DPIM Reduced Collagen Accumulation in BDL-Induced Fibrosis
As a hallmark of fibrosis, the collagen deposition in the liver was assessed by the Sirius red staining. Extensive collagen fibers was observed in the BDL liver (Figure 2b), when compared with the sham control (Figure 2a), which had a minimal collagen stain mainly around the central veins and portal triads, as in the non-operated normal liver (not shown). Importantly, the collagen depositions were qualitatively reduced in the DPIM-treated livers (Figures 2c–e). Quantitative analysis showed that the Sirius red positive area was dose dependently decreased by two higher concentrations of DPIM, with the most impressive reduction of 82% (P<0.001) at 1 g/l, followed by a 60% reduction (P<0.001) at 0.75 g/l (Figure 2f). There was a trend for collagen reduction by 0.5 g/l of DPIM; however it did not reach a significant level. A reduction and normalization in both body weight and liver weight were also noted after DPIM treatment when compared with the sham control (Supplementary Table 1). We also confirmed in the sham mice that DPIM treatment itself (1 g/l, 4 weeks) did not cause any collagen build-up (Figure 2f) or liver injury as measured by serum level of ALT (data not shown).

Representative images and quantification of Sirius red staining of liver sections. Minimum collagen deposit was seen in the liver of sham-operated mice (a). In contrast, a significant area (9.2% of the total) was positive for the Sirius red in the BDL liver (b). Treatment of BDL mice with 0.5 g/l (c), 0.75 g/l (d), and 1 g/l (e) of DPIM clearly reduced the collagen accumulation. (f) Quantification of the Sirius red positive areas confirmed a significant, dose-dependent reduction starting at 0.75 g/l, with the most robust reduction of 82% at 1 g/l. Each treatment group consisted of 6–8 mice. *P<0.05, ****P<0.001. Scale bar=200μm.
To obtain molecular evidence for supporting the Sirius red stain results, the mRNA level for Col1α1 and TGFβ1, and the protein level for α-SMA in the livers of different treatment groups were measured by real-time PCR and western blot, respectively. As expected, the transcription of Col1α1 (Figure 3a) and TGFβ1 (Figure 3b) genes were inhibited by DPIM, particularly at 1 g/l concentration. At the protein level, α-SMA was also suppressed by DPIM as measured by western blot and quantification (Figure 3c) or by immunohistochemistry (IHC) (Figure 3d).

DPIM suppressed the expression of Col1a1, TGFβ-1, and 1α-SMA genes in liver. Relative transcript level in the liver for Col1a1 (a) and TGFβ-1 (b), as measured by real-time PCR, were dose dependently suppressed by DPIM treatment. The protein expression of α-SMA was also reduced by DPIM as measured by western blot and quantification (c) and immunohistochemistry (d). *P<0.05, **P<0.01, ****P<0.001. Scale bar=50μm.
DPIM Reduced Infiltrating Immune Cells in BDL Liver
Our in vitro experiments revealed that DPIM has the ability to significantly alter chemokine signaling in cultured HSCs. To test the hypothesis that the anti-fibrotic properties of DPIM could be in part mediated by suppressing the chronic inflammatory response to BDL, we first examined the H&E-stained liver sections (Figure 4a), and found numerous infiltrating immune cells around the sinusoidal areas in the BDL liver, when compared with the sham control. Notably, the number of infiltrating cells was reduced by DPIM treatment (Figure 4a). By using IHC, we confirmed that DPIM indeed reduced the tissue areas positive for CD68, a marker for the circulating macrophages and the resident Kupffer cells (Figures 4b and c). We further showed that the BDL-induced expression of MCP-1 was effectively attenuated by DPIM at the mRNA (Figure 4d) and the protein (Figure 4e) level. Additional IHC experiment using antibody against F4/80 antigen, which is specifically expressed in mature microphages and Kupffer cells, also confirmed the anti-inflammatory effect of DPIM (Supplementary Figures 4a and b). Finally, IHC data obtained with a pan-cytokeratin antibody also revealed that the BDL-induced cellular proliferation was inhibited by DPIM (Supplementary Figure 5).


DPIM suppressed inflammation. (a) Representative of images of H&E staining of liver sections revealed that the number of infiltrating immune cells (intensely positive for hematoxylin, dark blue) around the sinusoidal region of the BDL liver was reduced by DPIM treatment. Scale bar=200μm. (b) DPIM significantly decreased the CD68+ stain in IHC, and at a dose of 1 g/l (c) it reduced the CD68+ value to 16.3% of that in the BDL. DPIM also significantly suppressed the expression of monocyte chemotactic protein-1 (MCP-1) at the messenger (d) and the protein (e) level, respectively. *P<0.05, ****P<0.001.
DPIM Altered the Balance of Specific TIMP-1/MMPS
As our in vitro data (Supplementary Figures 1 and 2) suggested that DPIM can change the expression of TIMP-1 and several MMP genes, we analyzed the effect of DPIM on TIMP/MMP balance in mouse liver samples. The results showed that the level of TIMP-1, which was barely detectable in sham liver, was significantly elevated in the BDL liver. Critically, the BDL-induced TIMP-1 expression was significantly suppressed after DPIM treatment at the mRNA (Figure 5a) and the protein (Figure 5b) level. At the same time, we also found that the transcription of MMP-8 (collagenase) was enhanced by almost threefolds in the BDL liver treated with DPIM (0.75 or 1 g/l) group (Figure 5c). A significant increase in transcription was also seen for the MMP-2 and MMP-9 (gelatinases), respectively (Figures 5d and e). A gelatin zymography further verified that specific activities for MMP-2 and MMP-9 were enhanced by the DPIM treatment (Figures 6a–c). Surprisingly, the total MMP activities, as measured by using a fluorogenic peptide substrate, were actually decreased by DPIM treatment at both concentrations of 0.75 and 1 g/l (Figure 6d).

DPIM affected ECM-modifying enzymes TIMP-1 and MMPs. The BDL-induced TIMP-1 expression was attenuated by DPIM at the transcript (a) and the protein (b) level, respectively. In parallel, transcript levels for MMP-8 (c), MMP-2 (d), and MMP-9 (e) were increased in the DPIM-treated livers. *P<0.05, **P<0.01, ***P<0.005.

DPIM affected MMPs activity. Specific activity for MMP-2 and MMP-9 in the liver extracts was measured on a gelatin zymography (a), followed by quantification (b, c), respectively, whereas the total hepatic MMPs activities was quantified by using a fluorogenic peptide substrate (d). It is noted that DPIM specifically induced the activity of MMP-2 and MMP-9, but simultaneously attenuated the total MMPs activities in the BDL liver. *P<0.05, **P<0.01, ***P<0.005.
DPIM Did Not Significantly Augment Anti-oxidation nor Induced Apoptosis
As some IMSs could act as anti-oxidants in vitro,10 we sought to study the anti-oxidative property of DPIM in vivo by measuring the level of hepatic glutathione (GSH) and the ratio of GSH/GSSG. As shown in Figures 7a and b, BDL-depleted GSH by 20% and decreased the ratio of GSH/GSSG by 21%. DPIM treatment (0.75 or 1 g/l) did not significantly replenish the anti-oxidative deficiency induced by BDL. It is noted that treatment of sham mice with 1 g/l of DPIM continuously for 4 weeks did not alter the redox state either.

DPIM slightly compensated the loss of anti-oxidative power in DBL liver. Treatment with DPIM for 4 weeks only appeared to slightly increase the GSH level (a) and the GSH/GSSG ratio (b), but not statistically significant. *P<0.05. (c) DAPI staining showed that DPIM treatment reduced the number of infiltrating immune cell (with small nuclei, arrows). For reference, hepatocytes in the tissue sections have large and round nuclei (arrow heads). TUNEL staining (empty arrow heads) revealed limited apoptosis induced by DPIM treatment.
Our gene expression profiling data on cultured cells (Supplementary Figure S1) also suggested that DPIM may influence apoptosis signaling. To investigate this possibility in vivo as a means resolve fibrosis, we performed TUNEL staining (green color in Figure 7c) on liver tissue sections. The results showed minimal apoptotic events (empty arrow heads) in the DPIM or the BDL group. Nevertheless, DAPI staining (blue color in Figure 7c) of the liver sections reinforced an early observation (Figure 4a) that DPIM treatment reduced the number of infiltrating macrophages (arrows, Figure 7c).
DISCUSSION
As an early event, inflammation precedes and promotes fibrogenesis during chronic liver injury. Both innate and adaptive immune responses have important roles in liver fibrogenesis.14 The role of different inflammatory cell types in the liver fibrogenesis has been studied by selectively removal of certain cell types, such as macrophages,15 natural killer cells,16 and B cells.17 At the molecular level, specific genes contributing to inflammation were also implicated as mediators for fibrogenesis.3, 18, 19, 20 Although the relationship between inflammation and fibrosis is not always straightforward due to the complex cross-talks among different cells types and pathways, reducing inflammation and immune response has been recognized as one of the major approaches to combat fibrosis. For example, the gene-based dominate-negative form of MCP-1 has been shown to suppress activated HSC and reduce liver fibrosis in rats.4 In this report, we showed that DPIM treatment significantly suppressed the recruitment of macrophages and/or the proliferation of Kupffer cells into the portal areas (Figures 4a and 7c), which could be fueled by the inflammatory chemokine MCP-1 (Figures 4b and c) expressed and secreted by multiple cell types in the pathological foci. Consequently, a reduction in inflammatory response could result in an inhibition of cellular proliferation around the bile duct area (Supplementary Figure 4). In this aspect, our findings certainly lend support for using small anti-inflammatory compounds to inhibit fibrosis. In separate studies by others, the non-steroidal anti-inflammatory drug, Celecoxib, has been explored and yielded both promising and conflicting results for fibrosis in animal models.21, 22 Possible reasons for the conflicting reports may include factors such as inconsistence in animal fibrosis modeling and compound dosing scheme and duration, etc. Or simply, Celecoxib alone is not sufficiently potent to suppress fibrosis across the board.
In healthy liver, the ECM homeostasis is precisely maintained by an appropriate balance between MMPs and TIMPs. However, in the chronic liver injury, an increase in the expression of TIMP-1 partly contributed to the decreased turnover of ECM.7, 9 Therefore, strategies for blocking TIMP-1 had been explored to improve fibrosis. For example, antibody against TIMP-1 has been demonstrated to attenuate liver fibrosis in a CCl4 rat model,23 while others reported that an increased expression of MMPs led to fibrolysis in animal models.24, 25 In addition, a single gene delivery of MMP-8 with adenoviral vectors led to amelioration of fibrosis in both BDL and CCl4-induced fibrosis models.26 We discovered in the current study that DPIM not only suppressed the expression of TIMP-1 (Figures 5a and b), but also enhanced the expression of collagenase MMP-8 (Figure 5c). We also detected the enhanced expression (Figures 5d and e) and activity (Figure 6) of MMP-2 and MMP-9 (gelatinase) by DPIM. Early studies suggested that an increase in the expression and secretion in MMP-224 and MMP-927 from mesenchymal cells and hepatocytes alike during recovery phase to be beneficial to resolving fibrosis by fibrolysis. One interesting observation was that despite of the enhancement of several specific MMPs (MMP-8, MMP-2, and MMP-9) by DPIM, the total MMPs activities of the whole liver were actually decreased by DPIM (Figure 6d). It may be possible that induction of a specific MMP(s), rather than the total MMPs, could be more critical for resolving fibrosis. More future studies are required to test this hypothesis.
The role of MCP-1 in attracting infiltrating immune cells to the damaged liver was well documented, beside MCP-1, other chemokines had been implicated during fibrogenesis in the BDL model, such as CCL5, CXCL.28 A more recent report indicate that MCP-1 can activate and transdifferentiate portal fibroblasts to myofibroblasts via a distinct receptor other than CCR2,5 and induce collagen production in portal fibroblasts in a paracrine manner. These observations not only strengthen the role of inflammation in fibrogenesis, but also pinpoint MCP-1 as a key molecular link between the infiltration of immune cells and the production of collagen. Given the similarity in portal fibroblasts and HSCs, it is not difficult to speculate that MCP-1 could have a very similar role in the activation, transdifferentiation and collagen production in HSCs. Again this hypothesis needs to be tested in future mechanistic studies.
There is a possibility that the anti-fibrotic effect of DPIM was indirectly mediated through a potential hepatocyte protection function of the compound. To address this possibility, we treated BDL mice with three doses (0.5, 0.75, and 1.0 g/l) for 4 weeks and measured the serum ALT, AST, ALP, GGT, and bilirubin activities. As expected, BDL caused a significant liver damage as judged by an elevation in liver enzymes activities, when compared with that from the sham mice. Notably, BDL mice treated with DPIM did not have lower level of liver enzymes activities, suggesting lack of hepatocyte protection against the ongoing BDL insult by this compound (Supplementary Table 1).
Although anti-oxidative property has been observed for DBZIM in the HSCs,10 DPIM on the other hand only displayed a very marginal anti-oxidative benefit in the BDL liver (Figures 7a and b). This suggests that DPIM itself is a weak anti-oxidant in vivo, and instead it manifests its anti-fibrotic effects through other mechanisms, which we would propose to include at least the indirect effect of anti-inflammation and the more direct effect of ECM remodeling resulted from an alteration in the expression of TIMP-1/MMPs.
Although many successful preclinical experiments based on targeting a single signaling pathway have been reported, very few therapeutic candidates can progress through various stages of human clinical trials smoothly, partly due to the complex mutlifactorial nature of fibrogenesis pathology. Hence, our findings on DPIM with inhibitory on MCP-1 and TIMP-1 may provide a fresh venue for devising effective anti-fibrotic therapy.
References
Friedman SL . Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;134:1655–1669.
Marra F, DeFranco R, Grappone C, et al. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: correlation with monocyte infiltration. Am J Pathol 1998;152:423–430.
Seki E, de Minicis S, Inokuchi S, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology 2009;50:185–197.
Imamura M, Ogawa T, Sasaguri Y, et al. Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology 2005;128:138–146.
Kruglov EA, Nathanson RA, Nguyen T, et al. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am J Physiol Gastrointest Liver Physiol 2006;290:G765–G771.
Beaussier M, Wendum D, Schiffer E, et al. Prominent contribution of portal mesenchymal cells to liver fibrosis in ischemic and obstructive cholestatic injuries. Lab Invest 2007;87:292–303.
Kossakowska AE, Edwards DR, Lee SS, et al. Altered balance between matrix metalloproteinases and their inhibitors in experimental biliary fibrosis. Am J Pathol 1998;153:1895–1902.
Iimuro Y, Brenner DA . Matrix metalloproteinase gene delivery for liver fibrosis. Pharm Res 2008;25:249–258.
Hemmann S, Graf J, Roderfeld M, et al. Expression of MMPs and TIMPs in liver fibrosis—a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol 2007;46:955–975.
Zhang C, Ding Z, Suhaimi NA, et al. A class of imidazolium salts is anti-oxidative and anti-fibrotic in hepatic stellate cells. Free Radic Res 2009;43:899–912.
Chua SW, Vijayakumar P, Nissom PM, et al. A novel normalization method for effective removal of systematic variation in microarray data. Nucleic Acids Res 2006;34:e38.
Iimuro Y, Nishio T, Morimoto T, et al. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology 2003;124:445–458.
Neumann U, Kubota H, Frei K, et al. Characterization of Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, a fluorogenic substrate with increased specificity constants for collagenases and tumor necrosis factor converting enzyme. Anal Biochem 2004;328:166–173.
Henderson NC, Iredale JP . Liver fibrosis: cellular mechanisms of progression and resolution. Clin Sci (Lond) 2007;112:265–280.
Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 2005;115:56–65.
Radaeva S, Sun R, Jaruga B, et al. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006;130:435–452.
Novobrantseva TI, Majeau GR, Amatucci A, et al. Attenuated liver fibrosis in the absence of B cells. J Clin Invest 2005;115:3072–3082.
Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 2007;13:1324–1332.
Seki E, De Minicis S, Gwak GY, et al. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest 2009;119:1858–1870.
Seki E, Brenner DA . Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 2008;48:322–335.
Yu J, Hui AY, Chu ES, et al. The anti-inflammatory effect of celecoxib does not prevent liver fibrosis in bile duct-ligated rats. Liver Int 2009;29:25–36.
Paik YH, Kim JK, Lee JI, et al. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut 2009;58:1517–1527.
Parsons CJ, Bradford BU, Pan CQ, et al. Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology 2004;40:1106–1115.
Watanabe T, Niioka M, Ishikawa A, et al. Dynamic change of cells expressing MMP-2 mRNA and MT1-MMP mRNA in the recovery from liver fibrosis in the rat. J Hepatol 2001;35:465–473.
Sakaida I, Hironaka K, Kimura T, et al. Herbal medicine Sho-saiko-to (TJ-9) increases expression matrix metalloproteinases (MMPs) with reduced expression of tissue inhibitor of metalloproteinases (TIMPs) in rat stellate cell. Life Sci 2004;74:2251–2263.
Siller-Lopez F, Sandoval A, Salgado S, et al. Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology 2004;126:1122–1133; discussion 1949.
Roderfeld M, Weiskirchen R, Wagner S, et al. Inhibition of hepatic fibrogenesis by matrix metalloproteinase-9 mutants in mice. FASEB J 2006;20:444–454.
Sahin H, Trautwein C, Wasmuth HE . Functional role of chemokines in liver disease models. Nat Rev Gastroenterol Hepatol 2010;7:682–690.
Acknowledgements
This work was supported by the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Supplementary information
Rights and permissions
About this article
Cite this article
Ding, Z., Kng, Y., Yang, H. et al. An orally available small imidazolium salt ameliorates inflammation and fibrosis in a murine model of cholestasis. Lab Invest 91, 752–763 (2011). https://doi.org/10.1038/labinvest.2011.11
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2011.11

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}