
Abstract
The nicotinic acetylcholine receptor α1 (nAChRα1) was investigated as a potential proinflammatory molecule in the kidney, given a recent report that it is an alternative urokinase plasminogen activator (uPA) receptor, in addition to the classical receptor uPAR. Two animal models and in vitro monocyte studies were involved: (1) In an ApoE−/− mouse model of chronic kidney disease, glomerular-resident cells and monocytes/macrophages were identified as the primary cell types that express nAChRα1 during hypercholesterolemia/uninephrectomy-induced nephropathy. Silencing of the nAChR α1 gene for 4 months (6 months on Western diet) prevented the increases in renal monocyte chemoattractant protein-1 and osteopontin expression levels and F4/80+ macrophage infiltration compared with the nonsilenced mice. These changes were associated with significantly reduced transforming growth factor-β1 mRNA (50% decrease) and α smooth muscle actin-positive (αSMA+) myofibroblasts (90% decrease), better glomerular and tubular basement membranes (GBM/TBM) preservation (threefold less disintegration), and better renal function preservation (serum creatinine 40% lower) in the nAChRα1-silenced mice. The nAChRα1 silencing was also associated with significantly reduced renal tissue calcium deposition (78% decrease) and calpain-1 (but not calpain-2) activation (70% decrease). (2) The nAChRα1 was expressed in vitro by mouse monocyte cell line WEHI-274.1. The silencing of nAChRα1 significantly reduced both calpain-1 and -2 activities, and reduced the degradation of the calpain substrate talin. (3) To further explore the role of calpain-1 activity in hypercholesterolemic nephropathy, disease severities were compared in CAST−/−ApoE−/− (calpain overactive) mice and ApoE−/− mice fed with Western diet for 10 months (n=12). Macrophages were the main cell type of renal calpain-1 production in the model. The number of renal F4/80+ macrophages was 10-fold higher in the CAST−/−ApoE−/− mice (P<0.05), and was associated with a significantly higher level of αSMA+ cells, increased GBM/TBM destruction, and higher serum creatinine levels. Our studies suggest that the receptor nAChRα1 is an important regulator of calpain-1 activation and inflammation in the chronic hypercholesterolemic nephropathy. This new proinflammatory pathway may also be relevant to other disorders beyond hyperlipidemic nephropathy.
Similar content being viewed by others


Main
The prevalence of overweight and obese individuals has increased rapidly within the past two decades.1, 2 Subsequently, a greater number of individuals are at risk of developing hypertension, atherosclerosis, coronary artery disease, diabetes, lipoprotein abnormalities, and other organ dysfunction.3 In addition, recent literature has shown that chronic hyperlipidemia, especially hypercholesterolemia, has lipotoxic effects in the kidney, which lead to renal mesangial and epithelial cell injury, and subsequent fibrosis through a complex inflammatory pathway.4, 5, 6 The hallmark of hyperlipidemic chronic kidney disease (CKD) is a macrophage dominated inflammatory response (foam cell) that ultimately progresses to glomerulosclerosis. This process may be associated with mesangial expansion, prominent mesangiolysis, capillary microaneurysms, and finally sclerosis.7 The molecular mechanisms that mediate this series of pathological events in the hyperlipidemic CKD remains poorly understood.
Urokinase plasminogen activator (uPA) is a growth factor-like protein, which is produced in large quantities by the kidney. Furthermore, uPA and its classical receptor uPAR have been implicated in various inflammatory–fibrotic disorders, including CKD.8 Evidence suggests that the uPA/uPAR system regulates cellular responses during inflammation, angiogenesis, wound healing, and tumor metastasis.9, 10, 11, 12, 13 The muscle-type nicotinic acetylcholine receptor α1 (nAChRα1) has recently been identified as an alternative urokinase receptor. We previously reported that in a mouse obstructive uropathy model of CKD, the nAChRα1 expression is upregulated on interstitial fibroblasts during fibrogenesis. Silencing of the nAChRα1 gene led to significantly fewer interstitial α smooth muscle actin-positive (αSMA+) interstitial myofibroblasts, fewer proliferating fibroblasts, better preserved tubular cells, and reduced overall fibrosis severity.14 This study investigates whether nAChRα1 has a similar protective role in hyperlipidemia-associated CKD.
The activated nAChRα1 also functions as a ligand-gated ion channel, known to mediate signal transduction at the neuromuscular junction.15 In addition to muscle cells, the muscle-type nAChRα1 is also expressed in vascular endothelium, macrophages, mesangial cells, and fibroblasts.14, 15 Currently, there exist two known endogenous nAChRα1 ligands: acetylcholine and uPA. On ligation, the receptor changes its conformation and becomes permeable to both calcium and sodium ions. Our previous studies show that on uPA ligation, the muscle-type nAChRα1 initiates intracellular signaling by functioning as a calcium channel. Although specific downstream signaling pathways remain to be delineated, it is believed that calpains, a class of cytosolic calcium-dependent cysteine proteases, may be involved.16, 17 Recent studies suggest that calpain-1 mediates the inflammatory response by having a role in inflammatory cell migration and chemotaxis, as well as in the expression of proinflammatory cytokines and adhesion molecules.18, 19, 20 Although it has not yet been studied in any detail in CKD, increased calpain activity has been reported in experimental kidney disease models and is thought to be detrimental in immunity-mediated acute glomerulonephritis.17
This study was designed to determine whether calpain-1 activation is regulated by nAChRα1 in vivo and in vitro, and to test the hypothesis that the nAChRα1-calpain-1 axis participates in the pathogenesis of hypercholesterolemia-induced renal inflammation and fibrosis. Our findings indicate that the in vivo functional knockdown of nAChRα1 significantly attenuates calpain-1 activity and renal inflammation in an ApoE−/− mouse model of hypercholesterolemic CKD. Data from monocyte cultures further validate that the nAChRα1 is an important regulator for calpain activation. Furthermore, overactivation of calpain-1 in mice due to the absence of its endogenous inhibitor calpastatin exacerbates hypercholesterolemic nephropathy.
MATERIALS AND METHODS
Antibodies and cDNA Reagents
Antibodies used in this study and their sources are rat monoclonal antibody to nAChRα1 subunit (Covance, Berkeley, CA, USA); goat polyclonal antibody to nAChRα1, antibodies to monocyte chemoattractant protein-1 (MCP-1), and osteopontin (OPN; Santa Cruz Biotechnology, Santa Cruz, CA, USA); rat monoclonal antibodies to F4/80, CD11b (Serotec, Oxford, UK); EPO horseradish peroxidase-conjugated monoclonal antibody to αSMA (Dako Corp., Carpinteria, CA, USA); fluorescein isothiocyanate (FITC)-conjugated monoclonal antibody to β-actin, antibodies to calpain-1 (clone 15C10 and rabbit polyclone), monoclonal anti-talin antibody (clone 8d4) (Sigma-Aldrich, St Louis, MO, USA); and goat antibody to type IV collagen (Southern Biotechnology Associates, Birmingham, AL, USA). The cDNA reagents used in the in vitro and in vivo RNA interference (RNAi) studies are the nAChRα1-siRNA-expressing construct psir2 and the matched scramble RNA-expressing construct pscr that we previously described.14
Cell Culture and nAChRα1-RNAi Experiments
WEHI-274.1 cells, a mouse monocyte cell line that expresses CD11b antigen,21 were used as an in vitro model to determine whether monocytes/macrophages express nAChRα1, and to investigate the regulatory action of nAChRα1 on calpain-1 activation. Cells were routinely cultured at 37°C with 5% CO2 in complete Dulbecco's modified Eagle's medium (DMEM/F12, 1:1, pH 7.35–7.45) (Mediatech, Manassas, VA, USA) containing 10% fetal bovine serum (vol/vol) and 0.05 mM 2-mercaptoethanol. For the nAChRα1-RNAi experiments, the psir2 and pscr cDNAs were transiently transfected into the WEHI-274.1 cell cultures in 12-well plates using siPORT XP-1 transfection agent for 24 h (Ambion, Austin, TX, USA) according to the manufacturer's instruction. The cells were then cultivated in the complete DMEM/F12 plus G418 (200 μg/ml) for an additional 24 h (n=6 per group, two independent experiments). Cells were harvested by dislodging adherent cells by scraping and collecting floating cells into centrifuge tubes. Following centrifugation at 800 r.p.m. for 5 min, 10 μl of cell pellet diluted in normal saline from each well was used to make cell smears for evaluating nAChRα1 expression by immunocytochemistry. The remaining cell pellets were lysed in sample buffer (100 mM Tris (pH6.8), 20% glycerol, 0.75% β-mercaptoethanol, 10 mM EDTA). Calpain activity was evaluated using the cell lysate by a previously depicted casein zymography method22 and talin—a calpain substrate—western blot analyses.
Animals and Experimental Design
In vivo nAChRα1-RNAi study in ApoE −/− mice
ApoE-deficient mice were used in this study as the model of hyperlipidemic nephropathy, because renal injury in these mice has been well characterized previously.7 Female ApoE−/− mice on a C57BL/6J background were purchased from the Jackson Laboratories (Bar Harbor, ME, USA), and were fed with Western-type diet containing 21% fat and 0.15% cholesterol (TD88137; Harlan-Teklad Laboratories, Indianapolis, IN, USA) beginning at 8 weeks of age. To functionally knockdown renal nAChRα1 expression, RNAi intervention started from 2 months on the Western diet. Naked plasmid DNA that expresses hairpin nAChRα1-siRNA (psir2) or scramble RNA (pscr) was administered (n=12 per group) using an aorta–renal hydrodynamic protocol modified from a previous publication.23 Briefly, a surgery was performed under general anesthesia to fully expose abdominal aorta and left renal artery. The distal end of left renal artery was ligated and a loose thread loop was placed in the proximal end. Aortic blood flow was temporarily blocked at points above and below the two renal arteries. Following an instant injection of 100 μg DNA (psir2 or pscr) in 200 μl normal saline, the preset left renal artery loop was immediately tied, and the upper and lower aortic blocking points were subsequently released. The left kidney was removed and tissues stored for subsequent studies (as a baseline disease control for the mice).24 After the surgery, the mice were maintained on the Western diet for an additional 4 months before being killed by exsanguination under general anesthesia. The right kidney and serum samples were collected and stored for further analyses.
Calpain overactivity study in CAST −/− ApoE −/− mice versus ApoE −/− mice
To investigate the role of calpain activation as a downstream effecter of the nAChRα1 pathway in the progression of hyperlipidemic kidney injury, a study was designed to compare the degree of kidney injury in ApoE−/− mice and in ApoE−/− mice lacking calpastatin. The calpain activity was manipulated using calpastatin (the only endogenous calpain inhibitor) gene knockout (CAST−/−) mice cross-bred onto the ApoE−/− mice. Both strains were on an identical genetic background (96.3–98.2% C57BL/6J, 1.9–3.8% 129/Sv). The resulting heterozygote mice in the first generation were intercrossed. After five generations of mating, double homozygote (CAST−/−ApoE−/− or DK) breeding pairs were obtained. The ApoE−/− single knockout (SK) mice used in the following experiment were also derived from the first-generation heterozygote mice. As a result of the cross-breeding, the breeding pairs for DK and SK mice were on a similar genetic background. The genotypes of all mice were verified on tail DNA by PCR, using the methods provided by Dr Saido (for CAST genotyping) and the Jackson Laboratory (for ApoE genotyping).25 The DK and SK female mice were fed with the Western diet beginning at 5 weeks of age (n=12). After 10 months on the Western diet, mice were killed and organ tissues were harvested (24-h urine samples were collected in individual mice using metabolic cages before killing). Additional DK and SK female mice (n=4 each) were fed with normal chow during the course of the experiment to serve as ‘normal’ controls. To avoid gender as a potential confounding factor, only female mice were used in our studies. All animal studies were approved by the IACUC of Seattle Children's Research Institute.
Biochemical Parameters
Plasma cholesterol levels were measured with a colorimetric assay (Cholesterol/Cholesteroyl Ester Quantitation Kit; BioVision Laboratories, Mountain View, CA, USA). Serum creatinine concentration was determined using the Jaffe reaction method previously established by Butler.26 Blood urea nitrogen levels were assessed using the Urea Nitrogen Liquid Reagent (Kinetic Method) as per the manufacturer’s instructions (TECO Diagnostics, Anaheim, CA, USA). Urinary albumin was measured using the QuantiChrom BCG Albumin Assay Kit (BioAssay Systems, Hayward, CA, USA).
Northern Blotting
Total RNA was isolated from kidneys using Trizol reagent (Invitrogen Corporation, Carlsbad, CA, USA) according to the manufacturer's protocol. Northern blot analyses for αSMA, transforming growth factor-β1 (TGF-β1), MCP-1, and OPN were performed as previously described.13, 14, 24
Western Blotting and Coimmunoprecipitation
Western immunoblotting (IB) and coimmunoprecipitation (IP/IB) experiments were performed following standard protocols.27 Specifically, calpain-1 was pulled down from 80 μg kidney protein samples in a nonreducing buffer with the monoclonal antibody (15C10) specific for subunit p80. The samples were then separated by a 10% SDS-PAGE in a reducing condition. Blots were probed with the rabbit polyclonal antibody for calpain-1 (domain IV) and labeled with the AlexaFluor680-conjugated secondary antibody (Molecular Probe). For western blot loading controls, blots were probed with the FITC-conjugated anti-β-actin monoclonal antibody. The stained fluorescent intensities were scanned and analyzed with a Typhoon TRIO Variable Mode Imager (Amersham Biosciences).
Histopathology
Paraffin-embedded kidney sections were stained with primary antibodies to nAChRα1, αSMA, type IV collagen, MCP-1, and OPN, and were labeled using a standard ABC kit protocol (Vector Laboratories, Burlingame, CA, USA). Immunofluorescent (IF) staining for F4/80+ macrophages was performed on frozen renal sections, identified with the FITC-conjugated goat anti-rat IgG secondary antibody (Southern Biotechnology Associates). Sections lacking primary antibodies were run in parallel as negative controls. For nAChRα1-expressing macrophage double stains, F4/80 and nAChRα1 were identified with FITC (green) and AlexaFlour680 (red) fluorescence, respectively. For calpain-1-expressing macrophage double stains, F4/80 and calpain-1 were labeled with AlexFlour680 (red) and FITC (green) fluorescence, respectively. Renal lipid deposition levels were detected by Oil Red O staining as previously described.28 Tissue calcium levels were measured on paraffin-embedded, Von Kosa stained kidney sections.29
Morphometric Analyses
Histological images were captured using a SPOT digital camera, and the stained areas were quantified using the Image-Pro Plus software as we previously described (Media Cybernetics, Silver Spring, MD, USA).14, 30 For immunohistology (αSMA, F4/80, MCP-1, OPN, and type IV collagen), the staining in glomeruli and tubulointerstitium were evaluated separately. The results were expressed as a positive percentage of each area of interest (glomeruli or tubulointerstitium). Disruption of glomerular and tubular basement membranes (GBM/TBM) was evaluated by measuring the staining intensities of the type IV collagen component of the GBM/TBM in combination with the disappearance of a typical loop structure. For this purpose, a specific color intensity threshold derived from the type IV collagen staining on baseline or ‘normal’ kidneys was first defined by the software as a strong staining (representing the intact basement membrane component). Only the strongly stained areas were counted as positive in terms of the integrity of the GBM/TBM. For the Oil Red O and Von Kosa stains, the staining was not separately quantified by glomeruli and tubulointerstitium because of the small areas of positivity; and the results were expressed as a positive percentage of the renal section area.
Statistical Analyses
Data were analyzed using the Student's t-test (parametric data) or Mann–Whitney U-test (nonparametric data), and the null hypothesis rejected at a P-value <0.05 unless specified elsewhere. Results are presented as mean±1 s.d. unless stated otherwise.
RESULTS
The nAChRα1 Regulates Calpain-1 Activation in Kidneys of ApoE−/− Mice and in Mononuclear Cell Culture
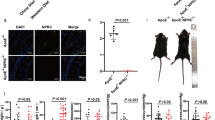
In baseline kidneys (2 months on Western diet, before DNA injection/uninephrectomy), renal nAChRα1 was barely detectable by western blotting in the ApoE−/− mice (Figure 1). After 6 months on a high fat diet, after DNA injection/uninephrectomy, the nAChRα1 protein expression was strongly and significantly upregulated in the kidneys of the control pscr-treated ApoE−/− mice. With gene silencing, the renal nAChRα1 expression was suppressed by 70% (psir2 versus pscr, n=8 per group, P<0.05). By immunolocalization (Figure 1), the nAChRα1 protein was mainly found in the diseased glomeruli and on tubulointerstitial cells. As illustrated by double IF staining, the majority of nAChRα1-expressing glomerular cells was identified as some F4/80+ infiltrated macrophages.

Renal nAChRα1 expression and silencing in the ApoE−/− mouse model of hypercholesterolemic CKD. (a) Western blot (WB) analysis for nAChRα1 shows a significant upregulation after uninephrectomy and 6-month Western diet, and a 70% gene knockdown by the nAChRα1-siRNA-expressing vector psir2. The β-actin bands were used to correct for protein loading. The histogram represents the relative band densities analyzed with the NIH image program (n=8). *P<0.05, pscr versus baseline or psir2. (b) The nAChRα1 immunohistochemical (IHC) staining detects a strong expression in the glomeruli and interstitium in the model of hyperlipidemic CKD and confirms the nAChRα1-silencing effect. At times, intensive glomerular nAChRα1 expression was associated with prominent mesangiolysis in the pscr-treated mice (as illustrated by the pscr, × 400). (c) Double IF staining illustrates infiltrated glomerular macrophages that coexpress (yellow overlap image) F4/80 (green) and nAChRα1 (red) in a diseased kidney of the pscr-treated ApoE−/− mouse. Original magnification: × 400.
The tissue calcium deposition levels were evaluated by a Von Kosa staining because the cell receptor nAChRα1 can function as a calcium channel, and changes in intracellular calcium concentration even though at micromol levels can regulate calpain-1 activation. Renal calcium levels were elevated to fivefold after 6 months of Western diet in the pscr-treated mice (0.75±0.6 versus 3.5±1.1%, at 6 months versus baseline, n=6, P<0.05). The nAChRα1-interference reduced the calcium level in the diseased kidneys by more than fourfold (0.79±0.6 versus 3.5±1.1%, psir2 versus pscr, n=6, P<0.05). The level of renal calpain-1 activity increased 10-fold compared with baseline levels in the pscr-treated mice (6 months versus baseline, n=8, P<0.05) (Figure 2a). The knockdown of the nAChRα1 for 4 months was associated with a 70% reduction in the active calpain-1 (58 kDa) level in the kidneys of the psir2-treated mice (psir2 versus pscr, n=8, P<0.05) (Figure 2a). No difference was found in renal activated calpain-2 protein in the psir2-treated compared with the pscr-treated mice (data not shown), despite significantly different calpain-2 activities found in monocyte cultures (Figure 2b). By immunohistochemistry, renal calpain-1 protein was expressed by glomerular and interstitial cells in the pscr-treated mice, but almost undetectable in the psir2-treated mice (Figure 2a).


The nAChRα1 is a key regulator for cellular calpain-1 activation. (a) Coimmunoprecipitation (IP/IB) studies (details shown in the Materials and method) show significantly less amount of the activated calpain-1 protein (58 kDa) in kidneys of the psir2-treated mice compared with the pscr-treated mice (n=6). The antibody reacts with bands at 80 kDa (latent), 58 kDa (activated and functional, indicated by arrowhead), and a series of smaller further degraded products of unknown biological significance. The graph shows the mean active bands densities ±1 s.d., corrected for protein loading using β-actin protein band densities. *P<0.05, pscr versus baseline or psir2. Calpain-1 immunohistochemical (IHC) staining detects clear expression in the glomeruli (2) and interstitium (1) in a pscr-treated mouse kidney. The glomerular and interstitial expression has largely disappeared in a psir2-treated mouse kidney (3). Original magnification: × 250. In (b), WEHI-274.1 monocytes express nAChRα1 in cultures as shown by IHC (1). A cell strongly expressing nAChRα1 is highlighted in (2). The nAChRα1 expression was effectively silenced by transient transfection of the psir2 vector (3). Original magnifications: (1) and (3), × 250; (2) × 400. The nAChRα1 silencing in the monocytes is associated with significantly reduced calpain-1 and -2 activities (zymogram), and reduced calpain substrate talin cleavage (WB). Talin protein was fully cleaved into the 190-kDa fragment in the pscr-transfected cells; significantly less amount of talin was cleaved in the psir2-transfected cells 48 h following RNAi. Intact talin=225 kDa. The histogram represents the relative band densities, analyzed with the NIH image program. *P<0.05, psir2 versus pscr, n=4 each.
In vitro the nAChRα1 was expressed by the mouse monocyte cell line WEHI-274.1. Silencing nAChRα1 expression on the cells significantly reduced the activities of both calpain-1 (60% decrease) and calpain-2 (90% decrease), and reduced the cleaving of calpain substrate talin by 20% (psir2 versus pscr, n=4, all P<0.05) (Figure 2b). These results suggest that the receptor nAChRα1 is an important upstream regulator of the calcium-dependent activation of calpain-1 in the hypercholesterolemic model of CKD.
Renal Macrophage Inflammation is Reduced in the nAChRα1-Silenced Mice
F4/80+ renal monocytes/macrophages, the dominant inflammatory cell type during hypercholesterolemic nephropathy, were significantly increased over time in the pscr-treated mice (Figure 3a). The numbers of F4/80+ cells were 2- and 10-fold elevated in the glomeruli and cortical tubolointerstitium, respectively, in the kidneys of the pscr-treated ApoE−/− mice (6 months versus baseline, n=6, P<0.05). The nAChRα1 knockdown by the psir2 treatment prevented the increase of macrophage infiltration in both the glomerular and tubulointerstitial compartments (6 months versus baseline, n=6, P>0.05). The depressed macrophage recruitment was coupled with reduced expression of two important monocyte chemoattractant chemokine genes: the MCP-1 and OPN. The baseline renal levels of MCP-1 and OPN mRNA and protein were minimal; expression was limited to the vascular walls and the interstitial macrophage-like cells (Figure 3b). In the pscr-treated mice, the mRNA and protein expression of the two chemokines significantly increased by three- to fivefold in all compartments of the kidney (6 months versus baseline, n=5, all P<0.05). In the nAChRα1-silenced mice, MCP-1 and OPN mRNA and protein levels remained very low at 6 months after being fed with Western diet (6 months versus baseline, n=5, mRNA or protein, all P>0.05). These finding suggest that the activation of receptor nAChRα1 is strongly associated with the regulation of macrophage recruitment in the model of hypercholesterolemic nephropathy.


The nAChRα1-RNAi reduces renal macrophage recruitment. (a) F4/80 IF stain shows a significantly greater presence of macrophages in both the glomeruli and in the tubulointerstitium after uninephrectomy and 6-month Western diet in the pscr-treated mice, but not in the psir2-treated mice. Using the Image-Pro Plus program, compartment-specific F4/80+ macrophages were quantified and results were expressed as a positive percent area of interest. #P<0.05, pscr versus baseline; *P<0.05, psir2 versus pscr, n=6. Original magnification: × 250. (b) Northern blot (NB) analysis illustrates a significant upregulation in MCP-1 and OPN mRNAs in the progression of hypercholesterolemic CKD seen in the pscr-treated mice 6 months after being fed on Western diet, but not in the psir2-treated mice (n=5). The histogram below the NB represents semiquantitative results (mean±s.d.) of NB analysis using the NIH image analysis program. The lower glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA bands were used to correct for RNA loading. IHC photomicrographs illustrate differences in the expression of kidney MCP-1 and OPN in mice treated with the nAChRα1 silencing (psir2) and scrambled (pscr) siRNA. The graph below summarizes the results of computer-assisted image analysis of the MCP-1+ or OPN+ glomerular and interstitial areas (n=5), confirming the findings of the NB analysis.
Early Profibrotic Renal Response is Blunted in the nAChRα1-Silenced Kidney
The hyperlipidemic nephropathy was characterized by a profibrotic renal response with upregulation of TGF-β mRNA (sixfold) in the pscr-treated mice (6 months versus baseline, n=5, P<0.05) (Figure 4). The renal TGF-β1 mRNA level was 50% lower in the nAChRα1-silenced mice compared with the pscr-treated mice (n=5, P<0.05) (Figure 4a). In baseline kidneys, no αSMA staining was found in glomerular tufts or tubulointerstitium except in the smooth muscle of vascular walls (Figure 4b). In the pscr-treated group, there was a strong de novo presentation of the αSMA+ myofibroblasts in both the glomeruli and tubulointerstitium after 6 months on Western diet. In the psir2-treated group, a small amount of glomerular αSMA+ staining was seen in the glomeruli and no tubulointerstitial myofibroblast was observed. As quantified by western blot analyses (Figure 3), the αSMA protein levels of the psir2-treated group were reduced by 90% when compared with the pscr-treated group (n=6, P<0.05). Northern blot analyses showed 50% reduced αSMA mRNA levels in the psir2-treated mice compared with the pscr-treated mice (n=5, P<0.05).

Reduced renal TGF-β1 mRNA and αSMA+ myofibroblasts by nAChRα1 silencing. (a) Kidney Northern blot analysis illustrates a significant twofold reduction in αSMA or TGF-β1 mRNA because of nAChRα1 silencing seen in the psir2-treated hyperlipidemic CKD mice, but not in the pscr-treated mice. The histogram represents semiquantitative results (mean±s.d.) of Northern blot analysis. The GAPDH mRNA bands were used to correct for RNA loading. #P<0.05, pscr versus baseline, *P<0.05, psir2 versus pscr, n=5. (b) αSMA IHC photomicrographs illustrate that αSMA+ myofibroblasts were absent in both the glomeruli and interstitium (excluding vascular smooth muscle) of the baseline kidneys. De novo expression of αSMA was observed in both glomerular and interstitial compartments 6 months after Western diet, but to a notably milder extent in the nAChRα1-silenced mice. Original magnification: × 400. Renal αSMA western blot analysis illustrates a significant 90% reduction in total αSMA protein in the psir2-treated mice compared with the pscr-treated mice (*P<0.05, n=6). The β-actin bands were used to correct for protein loading. The histogram represents mean band densities.
Less Severe Basement Membrane Damage and Better Renal Functions in the nAChRα1-Silenced Mice
The chronic inflammatory–fibrotic responses in this hypercholesterolemic CKD model led to a destructive process that was characterized by the damage of vascular, GBM/TBM, renal lipid deposition, and functional loss (Figure 5). As illustrated in Figure 5a, the baseline kidney demonstrated intensive collagen type IV staining of the basement membrane structures with intact capillary loops in the glomerular tufts/peritubular areas, and morphologically normal tubules. By 6 months, the area of the kidney occupied by the main basement membrane component collagen type IV was greatly reduced in the pscr-treated mice (three- to fivefold lower, 6 months versus baseline, n=6, P<0.05). The regular capillary loop structure appeared to be damaged and distorted, and some renal tubules were dilated. The collagen type IV component was significantly less damaged in the kidneys of the psir2-treated mice (two- to threefold higher, psir2 versus pscr, n=6, P<0.05), resulting in a better morphology of the kidneys (Figure 5a).

The nAChRα1 silencing protects basement membrane integrity and reduces lipid deposition in the kidney. (a) Type IV collagen IHC staining detects a strong expression in GBM/TBM and interstitial capillaries with clear loop-like structures in the baseline kidneys. The staining was significantly weakened in the progression of the hypercholesterolemic CKD, and was associated with the disappearance of the glomerular and interstitial loop structures and tubular dilation in the pscr-treated mice. Changes in the staining intensity and pathological morphology were less severe in the psir2-treated mice compared with the pscr-treated mice, indicating that nAChRα1 silencing protects GBM/TBM from disintegration. *P<0.05, pscr versus baseline or psir2, n=6. (b) Oil Red O stain photomicrographs illustrate differences in the level of kidney lipid deposition in mice treated with the nAChRα1 silencing (psir2) and scrambled (pscr) siRNA. The graph summarizes the results of computer-assisted image analysis of the lipid-stained area, showing a significant 98% reduction of lipid uptake by the nAChRα1-silenced group versus the scrambled group (n=6). Original magnification: × 400.
Renal lipid deposition, as measured by Oil Red O staining, was minimal at the baseline (Figure 5b). By 6 months, the Oil Red O stain-positive area was greatly increased by 15-fold in the kidneys of pscr-treated mice (6 months versus baseline, n=6, P<0.05). The lipid uptake and deposition in glomerular and tubulointerstitial tissues was significantly reduced in the nAChRα1-silenced mice (98% lower, psir2 versus pscr, n=6, P<0.05), despite similar serum cholesterol levels (569.1±26.2 versus 549.4±12.4 mg per 100 ml, psir2 versus pscr, n=5, P>0.05). In association with less severe renal pathological changes, the kidney function was better preserved in the psir2-treated mice (serum creatinine (SCr), 1.95±0.5 versus 3.2±0.7 mg per 100 ml, psir2 versus pscr, n=5, P<0.05).
Increased Endogenous Calpain Activity Promotes Renal Macrophage Inflammation in the CAST−/−ApoE−/− Mice
In the absence of calpastatin (the only known endogenous calpain inhibitor), renal levels of the activated calpain-1 were fivefold higher in the CAST−/−ApoE−/− (DK) mice compared with the ApoE−/− (SK) mice even on normal diet (n=4, P<0.05) (Figure 6a). Maintained on a Western diet for 10 months, the renal calpain-1 activation was significantly augmented by three- and eightfold in the DK and SK mice, respectively (Western diet versus normal diet, n=4, both P<0.05). The serum or kidney levels of the activated calpain-1 were about twofold higher in the DK mice compared with the SK mice (n=4, both P<0.05) 10 months on Western diet (Figure 6a). The integrin-to-cytoskeleton anchorage protein talin (a calpain substrate that is thought to be involved in the activation and migration of CD11b+ leukocytes/macrophages)31 was abundantly expressed—both the intact (225 kDa) and the calpain-cleaved (190 kDa) forms—in the kidneys of the SK mice on normal diet (Figure 6b). In the DK mice, all talin protein detected by western blot was in the calpain-cleaved form and in a significantly smaller quantity (77% decrease, DK versus SK, n=4, normal diet). The talin protein was completely cleaved and degraded to a barely detectable level in both the DK and SK mice 10 months after being fed with Western diet.

Calpain-1 activity was enhanced in kidneys of the CAST−/−ApoE−/− mice. (a) Western blot analysis shows significantly higher serum and renal levels of the active form calpain-1 (58 kDa, indicated by arrowheads) in the CAST−/−ApoE−/− double-knockout (DK) mice compared with the ApoE−/− SK mice after 10-month Western diet or normal chow. The albumin or β-actin bands were used to correct for protein loading. The histogram represents the relative band densities (mean±s.d., n=4). (b) Kidney talin western blot illustrates significantly lower levels in the DK mice versus SK mice on normal diet, indicative of calpain hyperactivity of the DK mice. Kidney talin was fully cleaved into the 190-kDa fragment (lower arrow), and was almost completely degraded into the antibody-undetectable products 10 months after Western diet. Intact talin=225 kDa (upper arrow). The histogram represents the relative band densities (n=4), analyzed with the NIH image program.
F4/80+ macrophages were rarely seen in the glomeruli and tubulointerstitium of the SK mice that were fed a normal diet (at the age of 11 months). F4/80+ cells were increased in the glomeruli and tubulointerstitium of the same-aged mice that had been on the Western diet for 10 months (Figure 7a). Associated with calpain-1 overactivity in the DK mice, a significantly higher baseline number of the F4/80+ macrophages was present in kidneys of the DK mice on normal diet (threefold higher, DK versus SK, n=4, P<0.05). Renal macrophages recruitment was enhanced by the Western diet in the DK mice (17- and 8-fold increases, in the glomeruli and tubulointerstitium, respectively, Western diet (n=6) versus normal diet (n=4), both P<0.05). This resulted in a significantly greater F4/80+ macrophage infiltration of the kidneys of the DK mice compared with the SK mice on the Western diet (10- and 3-fold higher, in the glomeruli and tubulointerstitium, respectively, n=6, both P<0.05) (Figure 7a). The difference in the degrees of the renal inflammatory response following the Western diet was further confirmed by western analysis of the leukocyte/macrophage marker CD11b (2.6-fold higher in the DK mice versus SK mice, n=4, P<0.05) (Supplementary Figure 1). As illustrated by IF double staining of a hypercholesterolemic DK kidney, all F4/80+ macrophages strongly expressed calpain-1 protein, and were the main source of calpain-1 in the inflamed kidney (Figure 7b).


Calpain overactivity promotes renal macrophage infiltration. (a) By IF stain, kidney F4/80+ macrophage infiltration was significantly exacerbated by calpain hyperactivity of the DK mice on either normal chow (n=4) or Western diet (n=6). Using the Image-Pro Plus program, compartment-specific F4/80+ macrophages were quantified and results were expressed as a positive percent area of interest. *P<0.05, DK versus SK, glomeruli or tubulointerstitium. Original magnification: × 250. (b) Double IF staining illustrates infiltrated glomerular macrophages that coexpress (yellow overlap image) F4/80 (red) and calpain-1 (green) in a diseased kidney of the DK mouse fed with high-fat diet for 10 months. Original magnification: × 400.
Calpain Hyperactivity Leads to More Severe Inflammatory Destruction and Renal Functional Loss
Intact GBM/TBM structures were clearly demonstrated in baseline kidneys, with no differences in the percentage area of collagen type IV staining between the DK and SK mice. Additional collagen type IV-positive staining appeared in the interstitial compartments in between the basement membrane structures (GBM/TBM) in the Western diet group of SK mice (Figure 8a), perhaps a manifestation of the fibrotic response of the model. This resulted in a slightly increased collagen type IV-stained area in the SK mice fed on Western diet (1.5-fold increase, Western diet versus normal diet, n=4, P<0.05). In contrast, the type IV collagen component of the GBM and TBM was significantly reduced by about 70 and 50%, respectively, in the Western diet group of DK mice (Western diet (n=6) versus normal diet (n=4), both P<0.05). The more severe GBM/TBM damage in the DK mice coincided with a stronger αSMA+ myofibroblast response incited by the Western diet (Figure 8b). The de novo appearance of glomerular αSMA+ myofibroblasts was fourfold higher in the DK mice when fed with the Western diet (DK versus SK, n=6, P<0.05). Tubulointerstitial αSMA+ area was also fivefold higher in the DK mice, but did not reach statistical significance (DK versus SK, n=6, P=0.06). These data suggest that calpain-1 hyperactivity promotes renal inflammatory–fibrotic cellular responses and GBM/TBM damage in the hypercholesterolemic mice.


Calpain overactivity accelerates kidney basement membrane destruction and myofibroblast transformation. Type IV collagen IHC staining (a) illustrates clear loop-like structures of renal basement membranes in mice fed normal chow (data of DK and SK mice were statistically similar and thus pooled). The type IV collagen expression in SK Western diet fed mice was intensified, with notable new staining in the interstitial area, and was associated with some dilated tubules, perhaps as a result of the early fibrotic response. However, the type IV collagen component of the basement membranes in the DK Western diet-fed mice was significantly lower. The predominant GBM/TBM disruption in the DK mice resulted in greater kidney αSMA+ myofibroblast transformation compared with the SK mice (b). #P<0.05, SK versus normal diet, n=4; *P<0.05, DK versus SK (n=6) or normal diet (n=4). Original magnification: × 400.
In terms of clinical parameters, serum cholesterol levels were above 1000 mg per 100 ml, but similar in the DK and SK mice after 10 months on a Western diet (Table 1). The 24-h urine albumin was 3.5-fold higher in the DK mice (DK versus SK, n=8, P<0.05), consistent with the pathological changes. Kidney function, as measured by SCr and blood urea nitrogen levels, was diminished to a greater extent in the DK mice (DK versus SK, n=8, both P<0.05) (Table 1).
DISCUSSION
Hyperlipidemia is thought to accelerate the progression of kidney disease, but the mechanisms by which hyperlipidemia exerts its deleterious effect remains largely unclear.3, 4, 32 This study specifically addresses the role of the muscle type nicotinic receptor, nAChRα1, on the progression of hypercholesterolemic nephropathy. Long-term in vivo knockdown of the nAChRα1 gene is renoprotective in the mouse model of hypercholesterolemic CKD. The results suggest that nAChRα1 mediates the progressive nephropathic inflammatory response by functioning as a regulator of calpain-1 activation. This study has suggested that the nAChRα1-calpain-1 activation intracellular conduit is involved as a novel proinflammatory pathway in the hypercholesterolemia-induced CKD.
The nAChR family has been shown to serve as a ligand-gated ion channel capable of mediating a variety of physiological activities. They are made up of a group diverse isoforms, each consisting of the specific assembly of five polypeptide subunits (the α1–10, β1–4, γ, δ, and ɛ) in either a homomeric or heteromeric configuration.33, 34, 35 Although the muscle-type nAChR ((α1)2β1γδ/ɛ) was originally discovered at neuromuscular junctions, it has since been identified in a variety of other cell types ranging from endothelial cells, vascular smooth muscle cells, immune cells, fibroblasts, and cancer cells.36 We have recently identified the nAChRα1 as an alternative uPA receptor. It is evident that the distinct structural components of individual nAChR isoforms directly relate to the physiological activity that the receptor regulates. For instance, it is now widely accepted that the nAChRα7 has an anti-inflammatory role; it is expressed by tubular cells in the kidney ischemia/reperfusion injury model, and is renoprotective.37 We recently reported that the nAChRα1 isoform promotes renal fibrosis through a mechanism that involves myofibroblast phenotypic regulation.14 In the obstructive fibrotic kidney, the uPA rather than acetylcholine may be the dominant endogenous ligand for interstitial nAChRα1.
Dyslipidemia is considered as a common factor contributing to the severity of renal function loss in patients with preexisting CKD.38 The ApoE−/− mouse model of hyperlipidemic nephropathy has been well characterized and widely used for mechanistic and interventional studies.3, 4, 30, 39 Hyperlipidemia is thought to promote chronic macrophage inflammation associated with significant renal-damaging effects in this model. In this study, we report that nAChRα1 expression is increased in both glomerular and interstitial cells including macrophages, and suggest that downstream calpain-1 activation promotes renal inflammation and chronic kidney injury. The silencing of nAChRα1 gene was associated with a considerable reduction in renal calpain-1 activation levels. The downstream reduction in calpain-1 activity led to a less severe renal inflammatory response, as suggested by our findings in mice with genetically induced calpain-1 overactivity because of the deficiency in the endogenous calpain inhibitor calpastatin. Calpain hyperactivity promoted 10-fold higher renal F4/80+ macrophage infiltration in the CAST−/−ApoE−/− mice, leading to significantly increased αSMA+ myofibroblasts, increased GBM/TBM destruction, and extensive kidney function loss (Figure 9).

Schematic summary of the nAChRα1-calpain pathways involved in hypercholesterolemic CKD. The muscle-type nicotinic receptor (nAChRα1) has two known endogenous ligands: the uPA and acetylcholine. On ligation the receptor transforms into a calcium channel, which subsequently activates the intracellular calcium-dependent cysteine protease calpain-1. The activated calpain-1 promotes cell motility, perhaps by cleaving cytoskeleton–integrin linking proteins such as talin and liberating cell surface integrins (eg, CD11b/CD18). Calpain-1 hyperactivity significantly accelerates renal macrophage recruitment in the mouse model of hypercholesterolemic CKD. The persistently accumulated renal macrophages, through autocrine or interactions with kidney-resident cells (paracrine), produce various destructive molecules such as chemokines (eg, MCP-1 and OPN), cytokines (eg, TGF-β), and metalloproteinases (eg, MMP-2 and MMP-9). These molecules further enhance renal inflammation, degrade the collagen components of GBM/TBM, and promote the matrix-producing myofibroblasts by transforming mesangial cells and pericytes, or by activating interstitial fibroblasts. The nAChRα1-regulated calpain-1 activity may also be involved in the lipid-uptake process by macrophages and kidney-resident cells. These destructive events ultimately lead to significant clinical proteinuria and kidney function loss. MCP-1, macrophage chemoattractant protein-1; OPN, osteopontin; TGF-β, transforming growth factor-β; MMP, metalloproteinase; SCr, serum creatinine; BUN, blood urea nitrogen; GBM/TBM, glomerular and tubular basement membranes.
Calpains are intracellular calcium-activated, nonlysosomal cysteine proteases that cleave cytoskeletal and submembranous proteins. The calpain system consists of two ubiquitous forms of calpain (calpain-1 or μ-calapin and calpain-2 or m-calpain), a tissue specific calpain (n-calpain), and a single known endogenous calpain inhibitor (calpastatin). Calpains participate in a variety of cellular functions, including regulation of cytoskeletal structures, cell-cycle progression, and cell spreading, adhesion and migration.40, 41, 42, 43 For instance, with the use of in vivo experiments McDonald et al44 and Chatterjee et al45 showed that the calpain inhibitor, N-acetyl-Leu-Leu-norleucinal, protected multiple organs from failure following hemorrhagic shock and reduced renal injury induced by ischemia/reperfusion. We have further addressed in this study that renal calpain-1 activation is, at least in part, regulated by the nAChRα1 receptor, which promotes the macrophage inflammation and disease progression in the experimental model of hypercholesterolemic CKD. The changes in calpain-1 activity, either downregulated by nAChRα1 silencing, or upregulated by calpastatin deficiency, respectively improved or intensified the macrophage inflammatory response and overall kidney damage. Previous studies suggest that calpain-1 regulates the inflammatory response by promoting inflammatory cell migration and proinflammatory cytokines production.18, 19, 20 Cell migration is a dynamic process that requires temporal and spatial regulation of integrin activation and focal adhesion assembly/disassembly. Talin is an actin and integrin tail-binding protein found in variety of tissues and cell types. Proteolysis of talin by calpain is essential for integrin activation, including leukocyte/macrophage CD11b/CD18 liberation and focal adhesion formation.30 Calpain-1 is also vitally involved in endocytosis;46 the cellular process macrophages and other types of cells use to uptake lipids.
Myofibroblasts are characterized as fibroblasts that have acquired myocyte characteristics such as contractile properties and αSMA expression. They are thought to be the primary source of the scar-forming extracellular matrix proteins in many solid organs, including kidneys. The density of glomerular or interstitial myofibroblasts is highly correlated with the decline of kidney functions in patients with CKD.47 The de novo appearance of glomerular αSMA+ cells is thought to be due to a transformation of activated mesangial cells.48 Interstitial myofibroblasts are derived from either activated peritubular/perivascular fibroblasts, or transformed pericytes and tubular epithelial cells.30 Pericytes are considered tissue-specific mesenchymal stem cells, with the potential to transdifferentiate into other functional mesenchymal cell types, such as the collagen-synthesizing myofibroblasts.49 It has been argued recently, by a fate-tracing study,50 that pericytes and perivascular fibroblasts rather than epithelial cells are the primary source of myofibroblasts in kidney fibrosis. The molecular basis of renal myofibroblast accumulation remains poorly understood. Growth factors such as TGF-β and members of the urokinase system have been shown to participate.8, 47 Disruption of the basement membrane is a key step in initiating the transformative process of glomerular or interstitial cells.51 This study has shown that cellular receptor nAChRα1 and its downstream activation of calpain-1 may be implicated in promoting the myofibroblast phenotypic transformation directly or indirectly, through TGF-β production and/or basement membrane destruction.24, 30, 51
Overall, the study demonstrates a unique relationship between the muscle type nicotinic receptor, nAChRα1, and the downstream activation of calpain-1 on the development of the hypercholesterolemia-induced nephropathic inflammatory response. Developing a more thorough understanding of the inflammatory-promoting mechanism by which the nAchRα1-calpain-1 activation conduit acts will better our understanding of CKD, and could lead to more targeted anti-inflammatory pharmaceuticals. In addition, as both the receptor and calpain-1 are involved in other disease pathologies, continued research on this new inflammatory-promoting pathway will be relevant to inflammatory disorders beyond the scope of CKD.
References
Ogden CL, Carroll MD, Curtin LR, et al. Prevalence of overweight and obesity in the United States 1999–2004. J Am Med Assoc 2006;295:1549–1555.
Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among United States adults 1999–2000. J Am Med Assoc 2002;288:1723–1727.
Wahba IM, Mak RH . Obesity and obesity-initiated metabolic syndrome: mechanistic links to chronic kidney disease. Clin J Am Soc Nephrol 2007;2:550–562.
Weinberg JM . Lipotoxicity. Kidney Int 2006;70:1560–1566.
Moorhead JF, Chan MK, El-Nahas AM, et al. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet 1982;2:1309–1311.
Ruan XZ, Moorhead JF, Fernando R, et al. Regulation in lipoprotein trafficking in the kidney: role of inflammatory mediators and transcription factors. Biochem Soc Trans 2004;32:88–91.
Wen M, Segerer S, Dantas M, et al. Renal injury in apolipoprotein E-deficient mice. Lab Invest 2002;82:999–1006.
Zhang G, Eddy AA . Urokinase and its receptors in chronic kidney disease. Front Biosci 2008;13:5462–5478.
Preissner KT, Kanse SM, May AE . Urokinase receptor: a molecular organizer in cellular communication. Curr Opin Cell Biol 2000;12:621–628.
Khatib AM, Nip J, Fallavollita L, et al. Regulation of urokinase plasminogen activator/plasmin-mediated invasion of melanoma cells by the integrin vitronectin receptor αVβ3. Int J Cancer 2001;91:300–308.
Preissner KT, Kanse SM, Chavakis T, et al. The dual role of the urokinase receptor system in pericellular proteolysis and cell adhesion: implications for cardiovascular function. Basic Res Cardiol 1999;94:315–321.
Gyetko MR, Sud S, Kendall T, et al. Urokinase receptor-deficient mice have impaired neutrophil recruitment response to pulmonary Pseudomonas aeruginosa infection. J Immunol 2000;165:1513–1519.
Zhang G, Kim H, Cai X, et al. Urokinase receptor modulates cellular and angiogenic responses in obstructive nephropathy. J Am Soc Nephrol 2003;14:1234–1253.
Zhang G, Kernan KA, Thomas A, et al. A novel signaling pathway: fibroblast nicotinic receptor α1 binds urokinase and promotes renal fibrosis. J Biol Chem 2009;42:29050–29064.
Albuquerque EX, Pereira EFR, Alkondon M, et al. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 2009;89:73–120.
Suzuki K, Kawabata Y, Sorimachi H . Structure, activation and biology of calpain. Diabetes 2004;53:S12–S18.
Peltier J, Bellocq A, Perez J, et al. Calpain activation and secretion promote glomerular injury in experimental glomerulonephritis: evidence from calpastatin-transgenic mice. J Am Soc Nephrol 2006;17:3415–3423.
Lokuta MA, Nuzzi PA, Huttenlocher A . Calpain regulates neutrophil chemotaxis. Proc Natl Acad Sci USA 2003;100:4006–4011.
Kavita U, Mizel SB . Differential sensitivity to interleukin-1 alpha and beta precursor proteins to cleavage by calpain, a calcium-dependent protease. J Biol Chem 1995;270:27758–27765.
Bellocq A, Doublier S, Suberville S, et al. Somatostatin increases glucocorticoid binding and signaling in macrophages by blocking the calpain-specific cleavage of Hsp 90. J Biol Chem 1999;274:36891–36896.
Boring L, Gosling J, Monteclaro FS, et al. Molecular cloning and functional expression of murine JE (monocyte chemoattractant protein 1) and murine macrophage inflammatory protein 1alpha receptors. J Biol Chem 1996;271:7551–7558.
Satish L, Blair HC, Glading A, et al. Interferon-inducible protein 9 (CXCL11)-induced cell motility in keratinocytes requires calcium flux-dependent activation of μ-calpain. Mol Cell Biol 2005;25:1922–1941.
Maruyama H, Higuchi N, Kameda S, et al. Sustained transgene expression in rat kidney with naked plasmid DNA and PCR-amplified DNA fragments. J Biochem 2005;137:373–380.
Zhang G, Kim H, Cai X, et al. Urokinase receptor deficiency accelerates renal fibrosis in obstructive nephropathy. J Am Soc Nephrol 2003;14:1254–1271.
Takano J, Tomioka M, Tsubuki S, et al. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J Biol Chem 2005;280:16175–16184.
Butler AR . The Jaffe reaction, identification of the colored species. Clin Chem Acta 1975;59:227.
Asanuma K, Yanagida-Asanuma E, Faul C, et al. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol 2006;8:485–491.
Jiang T, Liebman SE, Lucia MS, et al. Role of altered renal lipid metabolism and the sterol regulatory element binding proteins in the pathogenesis of age-related renal disease. Kidney Int 2005;68:2608–2620.
Mendoza FJ, Lopez I, Montes de Oca A, et al. Metabolic acidosis inhibits soft tissue calcification in uremic rats. Kidney Int 2008;73:407–414.
Zhang G, Kernan AK, Collins SJ, et al. Plasmin(ogen) promotes renal interstitial fibrosis by inducing epithelial to mesenchymal transition: roles of plasmin-activated signals. J Am Soc Nephrol 2007;18:846–859.
Anderson SI, Hotchin NA, Nash GB . Role of the cytoskeleton in rapid activation of CD11b/CD18 function and its subsequent downregulation in neutrophils. J Cell Sci 2000;113:2737–2745.
Bruneval P, Bariety J, Belair MF, et al. Mesangial expansion associated with glomerular endothelial cell activation and macrophage recruitment is developing in hyperlipidemic ApoE null mice. Nephrol Dial Transplant 2002;17:2099–2107.
Gotti C, Zoli M, Clementi F . Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci 2006;27:482–491.
Kalamida D, Poulas K, Avramopoulou V, et al. Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J 2007;274:3799–3845.
Lindstrom J, Anand R, Gerzanich V, et al. Structure and function of neuronal nicotinic acetylcholine receptors. Prog Brain Res 1996;109:125–137.
Cooke JP, Ghebremariam YT . Endothelial nicotinic acetylcholine receptors and angiogenesis. Trends Cardiovasc Med 2008;18:247–253.
Yeboah MM, Xiangying X, Javdan M, et al. Nicotinic acetylcholine receptor expression and regulation in the rat kidney after ischemia-reperfusion injury. Am J Physiol Renal Physiol 2008;295:F654–F661.
Muntner P, Coresh J, Smith C, et al. Plasma lipids and risk of developing renal dysfunction: the Atherosclerosis Risk in Communities Study. Kidney Int 2000;58:293–301.
Gervais M, Pons S, Nicoletti A, et al. Fluvastatin prevents renal dysfunction and vascular NO deficit in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2003;23:183–189.
Bronk SF, Gores GJ . pH-dependent nonlysosomal proteolysis contributes to lethal anoxic injury of rat hepatocytes. Am J Physiol 1993;204:G744–G751.
Schnellman RG, Yang X, Cross TJ . Calpains plays a critical role in renal proximal tubule (RPT) cell death. Can J Physiol Pharmacol 1994;72:602.
Trump BF, Berezesky IK . Calcium-mediated cell injury and cell death. FASEB J 1995;9:219–228.
Waters SL, Sarang SS, Wang KK, et al. Calpains mediate calcium and chloride influx during the late phase of cell injury. J Pharmacol Exp Ther 1997;283:1177–1184.
Chatterjee PK, Brown PA, Cuzzocrea S, et al. Calpain inhibitor-1 reduces renal ischemia/reperfusion injury in rat. Kidney Int 2001;59:2073–2083.
McDonald MC, Mota-Filipe H, Paul A, et al. Calpain inhibitor-1 reduces the activation of nuclear factor-kappa B and organ injury/dysfunction in hemorrhagic shock. FASEB J 2001;15:171–186.
Struewing IT, Barnett CD, Zhang W, et al. Frizzled-7 turnover at the plasma membrane is regulated by cell density and the Ca(2+)-dependent protease calpain-1. Exp Cell Res 2007;313:3526–3541.
Eddy AA, Neilson EG . Chronic kidney disease progression. J Am Soc Nephrol 2006;17:2964–2966.
Yabuki A, Mitani S, Fujiki M, et al. Comparative study of chronic kidney disease in dogs and cats: induction of myofibroblasts. Res Vet Sci 2010;88:294–299.
Paquet-Fifield S, Schlüter H, Li A, et al. A role for pericytes as microenvironmental regulators of human skin tissue regeneration. J Clin Invest 2009;119:2795–2806.
Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 2010;176:85–97.
Yang J, Liu Y . Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol 2001;159:1465–1475.
Acknowledgements
This work was supported by a Scientist Development grant (# 0730145N) from the American Heart Association National Center (GZ) and the Young Investigator Award from Seattle Children's Hospital and Regional Medical Center (GZ). We thank the RIKEN Brain Science Institute, Wako city, Saitama, Japan, for providing breading pairs of the CAST−/− mice and a PCR protocol for the genotyping. We thank Dr Renee C LeBoeuf (Director, NIH-funded Mouse Center for Metabolic Phenotype at University of Washington) for her generous support in collecting our preliminary data to initiate this study and for her insightful comments on the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Supplementary information
Rights and permissions
About this article
Cite this article
Zhang, G., Thomas, A., Marshall, A. et al. Nicotinic acetylcholine receptor α1 promotes calpain-1 activation and macrophage inflammation in hypercholesterolemic nephropathy. Lab Invest 91, 106–123 (2011). https://doi.org/10.1038/labinvest.2010.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2010.135

{kind=link}