
Abstract
Acetaminophen (APAP) induced increases in intrahepatic expression of interleukin (IL)-1α, IL-1β, and IL-1 receptor antagonist (IL-1ra), when administered intraperitoneally. These observations prompted us to define the pathophysiological roles of IL-1ra in APAP-induced liver injury. Compared with wild-type (WT) mouse-derived hepatocytes, IL-1ra-deficient (IL-1ra KO)-derived hepatocytes exhibited more resistance against APAP but not APAP-derived major toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI). Moreover, the amounts of a major APAP adduct (selenium-binding protein), an indicator of NAPQI generation from APAP, was significantly lower in IL-1ra KO mice than WT mice with depressed intrahepatic expression of CYP1A2, CYP2E1, and CYP3A11, the enzymes crucially involved in NAPQI generation from APAP. These observations would indicate that IL-1ra deficiency impaired APAP metabolism. IL-1α and IL-1β were expressed to similar extents in livers of untreated IL-1ra KO and WT mice. By contrast, the intranuclear amount of p65 of NF-κB, which can suppress the gene expression of CYP1A2, CYP2E1, and CYP3A11, was higher in untreated IL-1ra KO than WT mice. Moreover, when mice were intraperitoneally administered APAP (200 mg/kg), IL-1ra KO mice exhibited attenuated APAP-induced liver injury as evidenced by reductions in serum alanine transferase levels and histopathological changes such as centrilobular necrosis, hemorrhages, and leukocyte infiltration. Finally, when given 12 h before APAP challenge, IL-1α repressed the intrahepatic expression of CYP1A2, CYP2E1, and CYP3A11, eventually reducing APAP-induced liver injury, along with reduction in APAP adducts. Collectively, NF-κB was activated without any stimuli by the genetic disruption of IL-1ra, and suppressed cytochrome P450 enzyme expression, thereby reducing APAP-induced liver injury.
Similar content being viewed by others


Main
Acetaminophen (APAP), which is commonly used as an anti-analgesic and antipyretic, can be easily obtained over the counter. Easy access to this drug in the United States alone resulted in more than 100 000 APAP intoxication cases because of intentional or accidental intake, and about 500 fatal cases because of acute liver failure are reported annually.1 Although N-acetylcysteine, a precursor of glutathione (GSH), is used for the treatment of APAP-induced liver injury at present, clinical efficacy is insufficient because of its narrow therapeutic window.
Acetaminophen-induced toxicity is mediated by a metabolite, N-acetyl-p-benzoquinone imine (NAPQI) generated from APAP. Because NAPQI can react rapidly with sulfhydryl groups, it first reacts with GSH in hepatocytes and after depleting GSH, it then reacts with a number of intracellular proteins, thereby causing their dysfunction.2, 3, 4 Moreover, APAP hepatotoxicity is influenced further by several factors such as oxidative stress, inflammatory responses, cytokine production, and hepatocyte apoptosis.2, 3, 4 These factors contribute synergistically to damage. Indeed, we showed that inflammatory cytokines, chemokines, and leukocytes have some detrimental roles in the pathogenesis of APAP-induced liver injury.5, 6, 7
Interleukin-1 (IL-1), a pleiotropic proinflammatory cytokine, is produced by various kinds of cells such as neutrophils, macrophages, and fibroblasts. IL-1α and IL-1β, encoded by two distinct genes, show similar biological activities after binding to their specific and common receptor, type I IL-1 receptor (IL-1RI).8 IL-1 receptor antagonist (IL-1ra), exhibiting an identical β-pleated sheet structure as IL-1s,9, 10 can bind to IL-1R at the same sites with a similar affinity as IL-1s,11 but fails to associate with IL-1R accessory protein, which is indispensable for the biological activities of IL-1s.12 Thus, IL-1ra antagonizes IL-1 by binding to IL-1RI in a competitive manner. In various inflammatory diseases, such as rheumatoid arthritis and infectious diseases, IL-1ra is abundantly produced along with IL-1, probably to dampen the bioactivities of aberrantly produced IL-1s.11, 13 Mirroring its proinflammatory activity on IL-1, mice lacking IL-1ra (IL-1ra KO mice) spontaneously developed polyarthritis with increasing age.14 Consistently, we also observed that the lack of IL-1ra enhanced inflammatory responses during skin wound healing.15
In APAP-induced liver injury, intrahepatic IL-1 production was correlated with the magnitude of organ damage.4, 5, 6 Moreover, Blazka et al16, 17 demonstrated that the administration of anti-IL-1 antibody attenuated APAP-induced liver injury. Thus, we hypothesized that the genetic disruption of IL-1ra enhanced APAP-induced liver injury with aberrant inflammatory responses. Unexpectedly, APAP-induced liver injury was attenuated in IL-1ra KO mice, together with reduced generation of APAP adducts, the molecules involved crucially in APAP-induced liver injury, compared with wild-type (WT) mice. We further provided evidence to indicate that constitutive NF-κB activation in IL-1ra KO mice suppressed the intrahepatic expression of cytochrome P450 enzymes, which can generate NAPQI. Thus, the balance between IL-1s and IL-1ra expression in liver could affect the susceptibility of APAP-induced liver injury by modulating intrahepatic cytochrome P450 enzyme expression.
MATERIALS AND METHODS
Reagents and Antibodies
Acetaminophen and NAPQI were purchased from Sigma Chemical Company (St Louis, MO). The following monoclonal antibodies (mAbs) or polyclonal Abs (pAbs) were used in the present study: rat anti-mouse F4/80 mAb (Dainippon Pharmaceutical Company, Osaka, Japan), rabbit anti-myeloperoxidase (MPO) pAbs (Neomarkers, Fremont, CA), rabbit anti-APAP pAbs (Biogenesis Inc., Kingston, NH), rabbit anti-CYP1A2 pAbs, rabbit anti-CYP3A pAbs, rabbit anti-β-actin pAbs, rabbit anti-mouse NF-κB p65 pAbs (Santa Cruz Biotechnology, Inc, Santa Cruz, CA), rabbit anti-CYP2E1 pAbs (Daiichi, Pure Chemicals, Tokyo, Japan), and rabbit anti-single-strand DNA (ssDNA) pAbs (Dako, Kyoto, Japan). Recombinant murine IL-1ra and IL-1α proteins were prepared as described previously.18, 19
Mice
Pathogen-free 8- to 10-week-old male BALB/c mice were obtained from Sankyo Laboratories (Tokyo, Japan) and were designated as WT mice in the present experiments. Age- and sex-matched IL-1ra KO mice, backcrossed to BALB/c mice for more than eight generations, were used in the following experiments.15, 20 All mice were housed individually in cages under the specific pathogen-free conditions during the experiments. All animal experiments were approved by the Committee on Animal Care and Use in Wakayama Medical University.
Isolation and Culture of Mouse Hepatocytes
Mouse hepatocytes were isolated and cultured as describe previously.21 Briefly, calcium- and magnesium-free Hanks' solution supplemented with EGTA (0.5 mM) and Tris (25 mM, pH 7.4) was perfused through the liver via the portal vein for 15 min at 37°C The liver was then perfused with media-containing 0.05% collagenase (Gibco, Long Island, NY) at a flow rate of 5 ml/min for 20 min. Thereafter, the liver was removed, minced, and filtered. Hepatocytes were separated from nonparenchymal cells and debris by the centrifugation at 50 g for 1 min. Cell viability, as judged by Trypan blue exclusion, was usually >80%. Isolated hepatocytes were cultured onto a 96-well plate (2 × 104 cells per well) in Williams's medium E (Gibco) supplemented with 10% fetal bovine serum (Gibco), 100 U/ml penicillin/streptomycin and 10−7 M insulin at 37°C with 5% CO2. After 4 h incubation, cells were washed with PBS and then replaced with plain culture medium (control) or medium-containing APAP (5 mM) or NAPQI (400 μM). At 4 h later, cells were washed with PBS and then replaced with plain medium. The viability of hepatocytes was assessed by using a Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions. The data were expressed as percentage (%) of post-treatment to pretreatment viabilities. In some experiments, isolated hepatocytes from WT mice were cultured with rIL-1α (10 ng/ml) or rIL-1ra (40 ng/ml) for 6 h, and subjected to the following analyses.
APAP-Induced Liver Injury in Mice
In all experiments, mice were allowed free access to water but not food for 10 h before APAP challenge. APAP solution was made by dissolving the compound in phosphate-buffered saline (PBS, pH 7.2) and warmed to 37°C immediately before each experiment. Mice were intraperitoneally administered with APAP at the doses of 100–300 mg/kg in preliminary experiments. We observed that 200 mg/kg was the highest dose that did not cause any mortality in WT mice and therefore, we used this dose unless indicated otherwise. In some experiments, mice were intraperitoneally administrated with rIL-1α (1 μg/mouse), or rIL-1ra (40 μg/mouse) at 12 h before APAP challenge (200 mg/kg).
Determination of Serum Alanine Aminotransferase Levels
Whole-blood samples were collected at the indicated time intervals after APAP challenge to determine serum alanine transferase (ALT) levels with a Fuji DRI-CHEM 3500 V as instructed by the manufacturer (Fuji Medical System, Tokyo, Japan).
Histopathological Analysis
Liver tissues were obtained at the indicated time intervals after APAP challenge and were fixed in 4% formaldehyde buffered with PBS (pH 7.2) and then embedded with paraffin. Thereafter, sections (6 μm thick) were stained with hematoxylin and eosin. Pathological changes in the livers were evaluated by an examiner without a prior knowledge of the experimental procedures and scored as follows; 0, no necrosis; 1+, minimal centrilobular necrosis; 2+, severe necrosis but confined to centrilobular regions; 3+, necrosis extending from central zone to portal triads; 4+, massive necrosis spreading most regions.5, 6, 7
Enumeration of Infiltrating Inflammatory Cells and Apoptotic Hepatocytes
An immunohistochemical analysis was performed using anti-MPO pAbs for neutrophils, anti-F4/80 mAb for macrophages, and anti-ssDNA pAbs for apoptotic hepatocytes, as described previously.5, 6, 7 The numbers of infiltrating neutrophils, macrophages, and apoptotic hepatocytes in the liver were enumerated on 10 randomly chosen visual fields at × 400 magnification and the average of the 10 fields was calculated.7 All measurements were performed by an examiner without a prior knowledge of the experimental procedures.
Extraction of Total RNAs and Semiquantitative RT-PCR
Total RNAs was extracted from the removed liver tissues using ISOGEN (Nippon Gene, Toyama, Japan), followed by a semiquantitative RT-PCR analysis, as described previously.5, 6, 7 Briefly, 5 μg of total RNAs was reverse transcribed into cDNA using the Superscript II Reverse Transcriptase (Invitrogen Life Technologies, Tokyo, Japan) with Oligo (dT) Primers (Invitrogen Life Technologies). The resultant cDNA was amplified with Taq polymerase (Nippon Gene, Tokyo, Japan), using specific sets of primers with optimal cycles consisting of 94°C for 1 min, optimal annealing temperature for 1 min, and 72°C for 1 min, followed by the incubation at 72°C for 3 min (Table 1). The PCR products were then fractionated on a 2% agarose gel containing 0.3% ethidium bromide, and the band intensities were measured using NIH Image Analysis software version 1.63 (National Institutes of Health, Bethesda, MD). The ratio of each band to β-actin was determined. To standardize the condition of gel staining, a constant amount of control DNA marker was electrophoresed every time. The PCR procedure was performed at least three times for each sample.
Western Blotting
In some experiments, liver samples or hepatocytes (1 × 106) were homogenized with a lysis buffer (10 mM PBS, 1% NP-40, 4 mM EDTA) containing Complete Protease Inhibitor Cocktail (Roche Diagnostics, Tokyo, Japan) and centrifuged to obtain lysates. The lysates were electrophoresed in a 10% SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane. After the immersion with the blocking buffer (10 mM Tris-HCl, pH 7.4, containing 0.5 M NaCl, 5% skim milk, and 0.5% Tween-20), the membrane was incubated with anti-APAP pAbs, anti-CYP1A2 pAbs, anti-CYP2E1 pAbs, anti-CYP3A pAbs, or anti-β-actin pAbs at 4°C overnight. After the incubation with horseradish peroxidase-conjugated secondary Abs for 1 h at room temperature, the immune complexes were detected by using ECL-western blotting detection system (Amersham Bioscience Japan, Tokyo, Japan) according to the manufacturer's instructions. The band intensities were measured using NIH Image Analysis software version 1.63 (National Institutes of Health, Bethesda, MD). The ratio of each band to β-actin was determined.
Measurement of Intrahepatic Glutathione
Liver tissues were obtained at indicated time interval after APAP challenge and were homogenized in 5% metaphosphoric acid. The homogenates were centrifuged at 3000 g for 10 min. GSH levels in the supernatant were measured with a commercial GSH measurement kit (BIOXYTECH GSH400, OxisRsearch, Portland, OR) according to the manufacturer's instruction. The data were expressed as GSH (nmol)/liver weight (g).
Enzyme-Linked Immunosorbent Assay
Livers were obtained and homogenized with 0.3 ml PBS containing Complete Protease Inhibitor Cocktail (Roche Diagnostics), followed by three cycles of freezing and thawing. The homogenates were centrifuged at 10 000 g for 15 min. Supernatants were used to determine IL-1ra, IL-1α, IL-1β, IL-6, and IL-10 levels with enzyme-linked immunosorbent assay kits (IL-1ra, IL-1α, and IL-1β; R&D Systems, Minneapolis, IL-6; COSMO BIO, Tokyo, Japan, IL-10; Bender Medsystems Inc., Burlingame, CA) according to the manufacturer's instructions. The detection limits were as follows: IL-1ra, >7 pg/ml, IL-1α, >2.5 pg/ml, IL-1β, >3 pg/ml, IL-6, >3 pg/ml, and IL-10, >15 pg/ml. Total protein in the supernatant was measured with a commercial kit (BCA protein assay kit, Pierce, Rockford, IL), using BSA as a standard. The data were expressed as the target molecule (pg/ml)/total protein (mg/ml) for each sample.
Extraction of Nuclear Proteins and Measurement of NF-κB p65 Protein
Cell lysates from livers or hepatocytes (1 × 106) were further separated into cytosol and nuclear fractions using the NE-PER method (Pierce) as described previously.22 Each fraction was subjected to western blotting using anti-NF-κB p65 pAbs. In parallel, nuclear fractions were analyzed by using an EZ-Detect Transcription Factor kit (Pierce) to determine NF-κB p65 protein bound to the corresponding cis element, according to the manufacturer's instructions. The NF-κB activities were expressed as relative light units divided by total protein contents for each sample.
Determination of Blood APAP Concentrations by Gas Chromatography–Mass Spectrometry
At the indicated time intervals after APAP administration (200 mg/kg), the blood samples were collected from both strain mice. The blood samples were mixed with internal standard solution (o-acetamidephenol; 10 μg/10 μl) and 0.1 M borate sodium buffer (pH 10.5), followed by the adjustment of pH to 10 with 0.5 N sodium hydroxide solution. Then, the mixture was applied to an Extrelut NT® column (Merck), followed by the elution with ethyl acetate. The eluate was condensed and was converted to trimethylsilyl (TMS) derivatives with N,O-bis[trimethylsilyl]trifluoroacetamide +1% trimethylchlorosilane. The solution (1 μl) was applied onto a gas chromatography–mass spectrometry apparatus (A model Trace GC and MS system, Thermo Fisher Scientific, CA). Quantity of APAP was determined as the average area ratio of APAP-TMS (5.77 min, m/z 166 and 181) to o-acetamidephenol-TMS (5.10 min, m/z 166) as an internal standard.
Statistical Analysis
The means and s.e.m.s were calculated for all parameters determined in this study. Statistical significance was evaluated using analysis of variance or Mann–Whitney's U-test. P<0.05 was accepted as statistically significant.
RESULTS
Reduced Susceptibility of IL-1ra-Deficient Hepatocytes to APAP Toxicity
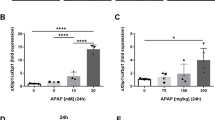
At first, we examined the susceptibility of cultured hepatocytes derived from WT and IL-1ra KO mice to APAP and its toxic metabolite, NAPQI. WT mouse-derived hepatocytes were more sensitive to APAP than IL-1ra KO mouse-derived ones, in terms of cell survival rates, although both hepatocytes showed similar sensitivities to NAPQI (Figure 1a). NAPQI, once generated from APAP, covalently binds to cysteine residues on proteins and induces the formation of 3-(cysteine-S-yl) APAP adducts, which is presumed to be responsible for APAP-induced hepatotoxicity. Thus, it is probable that there exist differences in the NAPQI and subsequent APAP adduct generation between WT and IL-1ra KO mice. To address this assumption, we determined the amount of the major APAP adduct (56 kDa selenium-binding protein) reflecting NAPQI amounts in the homogenates of hepatocytes and livers. After APAP addition to the culture medium, the amounts of APAP adducts were higher in WT-derived hepatocytes, than IL-1ra KO-derived ones (Figure 1b). In livers of untreated WT and IL-1ra KO mice, APAP adducts were barely detected, but the intraperitoneal injection of APAP (100–300 mg/kg) increased the amount of APAP adducts in a dose-dependent manner to a larger extent in WT mice than in IL-1ra KO ones (Figure 1c).23, 24 We next examined blood APAP concentration and intrahepatic GSH levels in both strain mice at 1 and 2 h after APAP challenge (200 mg/kg), because the half-life of APAP was 1-3 h in vivo.25 At the indicated time intervals, blood APAP levels was significantly higher in IL-1ra KO mice than in WT mice (Figure 1d). Intrahepatic GSH can detoxify NAPQI generated from APAP by the conjugation, but there was no significant difference in intrahepatic GSH level between unchallenged WT and IL-1ra KO mice. APAP challenge depleted intrahepatic GSH, to a similar extent, in both strain mice (Figure 1e). Thus, the absence of IL-1ra impaired NAPQI generation from APAP and the subsequent APAP adduct formation, eventually resulting in the attenuation of APAP hepatotoxicity.

(a) The susceptibility of hepatocytes isolated from WT and IL-1ra KO mice to APAP or NAPQI. Hepatocytes were isolated from WT and IL-1ra KO mice, and cultured in the presence of APAP (5 mM) or NAPQI (400 μM) for 4 h, as described in ‘Materials and Methods’. Each value represents mean±s.e.m. (n=6 animals). **P<0.01, WT vs IL-1ra KO mice. (b) Western blotting analysis of APAP adducts in hepatocyte homogenates from WT and IL-1ra KO mice after APAP treatment. Representative results from six independent experiments are shown here. (c) Western blotting analysis of APAP adducts in the livers of WT and IL-1ra KO mice administered with APAP (100–300 mg/kg). Representative results from six independent experiments are shown here. (d) Blood acetaminophen concentrations in WT and IL-1ra KO mice after APAP challenge (200 mg/kg). Each value represents mean±s.e.m. (n=6 animals). **P<0.01, WT vs IL-1ra KO mice. (e) Intrahepatic GSH levels in WT and IL-1ra KO mice after APAP challenge (200 mg/kg). Each value represents mean±s.e.m. (n=6 animals).
Intrahepatic Expression of Cytochrome P450 Enzymes in the Livers of WT and IL-1ra KO Mice Before and After APAP Challenge
Among cytochrome P450 enzymes, CYP1A2, CYP2E1, and CYP3A11 are presumed to be responsible for the generation of NAPQI from APAP.26, 27, 28, 29, 30 Hence, we next evaluated intrahepatic expression of these enzymes at both mRNA and protein levels in untreated and treated WT and IL-1ra KO mice. Before APAP challenge, IL-1ra KO mice exhibited reduced mRNA expression of these enzymes, compared with WT mice (Figure 2). APAP administration significantly decreased the gene expression of these CYPs in WT mice. Although APAP did have little effects on mRNA expression of these enzymes in IL-1ra KO mice, their gene expression levels were still lower in IL-1ra KO than in WT mice after APAP administration (except for CYP2E1 at 24 h; Figure 2). Concomitantly, IL-1ra KO mice exhibited less protein levels of these enzymes, compared with WT mice (Figure 3). These observations would indicate that IL-1ra deficiency reduced intrahepatic expression of CYP1A2, CYP2E1, and CYP3A11, the enzymes that could generate NAPQI, a toxic metabolite crucially involved in APAP-induced liver injury.

RT–PCR analysis for gene expression of CYP1A2, CYP2E1, and CYP3A11 in the livers of WT and IL-1ra KO mice before or after APAP treatment (200 mg/kg). Representative results from six independent experiments are shown in (a). The ratios of CYP1A2 (b), CYP2E1 (c), and CYP3A11 (d) to β-actin in the livers of WT and IL-1ra KO mice are shown here. Each value represents mean±s.e.m. (n=6 animals). *P<0.05, WT vs IL-1ra KO mice.

Western blotting analysis of CYP1A2, CYP2E1, and CYP3A in the livers of WT and IL-1ra KO mice before or after APAP treatment (200 mg/kg). Representative results from six independent experiments are shown in (a). The ratios of CYP1A2 (b), CYP2E1 (c), and CYP3A (d) to β-actin in the livers of WT and IL-1ra KO mice are shown in (d). Each value represents mean±s.e.m. (n=6 animals). *P<0.05, **P<0.01, WT vs IL-1ra KO mice.
Intrahepatic NF-κB Activation in WT and IL-1ra KO Mice Before and After APAP Challenge
Interleukin-1-mediated signal can activate NF-κB, a transcription factor that can suppress CYP gene transcription.31, 32, 33 Consistently, IL-1α treatment in WT-derived hepatocytes enhanced NF-κB activation and reciprocally attenuated the expression of CYP1A2, CYP2E1, and CYP3A11, whereas IL-1ra had little effect on NF-κB activation and subsequent expression of these enzymes (Figure 4). In spite of the absence of significant differences in intrahepatic IL-1α and IL-1β contents between untreated WT and IL-1ra KO mice, IL-1ra protein was constitutively expressed in the liver of WT mice (Table 2). This may mirror our observations that IL-1ra alone had little effects on IL-1α and IL-1β expression by WT-derived hepatocytes (our unpublished data). Thus, it is highly probable that NF-κB activation can be enhanced by deficiency of IL-1ra, a potent inhibitor of IL-1-mediated NF-κB activation. Indeed, nuclear NF-κB p65 protein levels were higher in IL-1ra KO mice than WT mice before APAP challenge, although there were no differences in cytosolic NF-κB p65 proteins between these two strains (Figure 5a and b). Concomitantly, the amount of NF-κB p65 bound to the corresponding cis element was significantly increased in lL-1ra KO mice, compared with WT mice (Figure 5c). These observations implied that, under the unstimulated condition, the disruption of IL-1ra resulted in constitutively enhanced NF-κB activation, which eventually led to suppressed expression of CYP1A2, CYP2E1, and CYP3A11. By contrast, APAP increased intrahepatic NF-κB-binding activity in WT mice but not IL-1ra KO ones.

(a) The effects of exogenous IL-1α and IL-1ra on DNA-binding activity of the nuclear NF-κB p65 in WT-derived hepatocytes. All values represent the mean±s.e.m. (n=6 independent experiments). *P<0.05 vs vehicle treatment. (b) Western blotting analysis of CYP1A2, CYP2E1, and CYP3A in WT-derived hepatocytes after IL-1α and IL-1ra treatment. Representative results from six independent experiments are shown here.

(a, b) Western blotting analysis of NF-κB p65 in cytosol and nuclear extracts of untreated WT and IL-1ra KO mice. Representative results from six independent experiments shown are in (a). The ratios of NF-κB p65 to β-actin were calculated and are shown in (b). All values represent the mean±s.e.m. (n=6 animals). *P<0.05, WT vs IL-1ra KO mice. (c) DNA-binding activity of the nuclear NF-κB p65 in the livers of WT and IL-1ra KO mice before and after APAP treatment (200 mg/kg). All values represent the mean±s.e.m. (n=6 animals). *P<0.05, WT vs IL-1ra KO mice.
Attenuated APAP-Induced Liver Injury in IL-1ra KO Mice After APAP Challenge
Reduced NAPQI generation from APAP in IL-1ra KO mice prompted us to examine APAP-induced liver injury in IL-1ra KO and WT mice. There were no apparent differences in serum ALT levels (WT vs IL-1ra KO: 39.0±2.6 vs 48.4±6.3 IU/l) and histopathological structure (data not shown) between unchallenged WT and IL-1ra KO mice. However, the administration of APAP (200 mg/kg) increased serum ALT levels in WT mice to a higher level than IL-1ra KO mice even at 2 h (WT vs IL-1ra KO: 399±123 vs 98.6±43.0 IU/l, P<0.01) and later thereafter (Figure 6a). Both WT and IL-1ra KO mice did not exhibit apparent histopathological changes such as centrilobular necrosis and leukocyte recruitment, at 2 h after APAP challenge (Figure 6b), but the numbers of apoptotic hepatocytes were higher in WT mice than IL-1ra KO mice (Figure 7). Later than 6 h after APAP challenge, WT mice showed more severe histopathological changes including hemorrhages and centrilobular necrosis than IL-1ra KO mice (Figure 6a–c). Moreover, APAP caused intrahepatic infiltration of both neutrophils and macrophages later than 6 h in WT mice to a larger extent than IL-1ra KO mice (Figure 8). We examined the intrahepatic gene expression of proinflammatory cytokines and chemokines in WT and IL-1ra KO mice. Under the used experimental conditions, the gene expression of IL-1α, IL-1β, CXCL1/KC, CXCL2/MIP-2, CCL2/MCP-1, and CCL3/MIP-1α was faintly detected in the livers of untreated WT and IL-1ra KO mice (Figure 9a). Later than 6 h APAP challenge, the gene expression of these cytokines and chemokines was augmented in WT mice to larger extents than IL-1ra KO mice (Figure 9b–g). By contrast, there was no significant difference in the intrahepatic expression of IL-6 and IL-10, the cytokines that had protective roles in APAP-induced liver injury (data not shown).34, 35 These observations would imply that impaired NAPQI generation in IL-1ra KO mice reduced the initiation of APAP-induced liver injury and subsequent inflammatory reactions as evidenced by reduced IL-1 and chemokine expression.

(a) Analysis of serum ALT levels in WT (n=10) and IL-1ra KO (n=10) mice at 2, 6, 10, and 24 h after APAP challenge. All values represent the mean±s.e.m. **P<0.01, WT vs IL-1ra KO mice. (b, c) Histopathological analysis on livers of WT and IL-1ra KO mice after APAP challenge (H&E staining, original magnification, × 200). Representative results from six independent experiments are shown in (b). Histological changes were scored as described in ‘Materials and Methods’ and shown in (c; n=6). All values represent the mean±s.e.m. **P<0.01, WT vs IL-1ra KO mice.

(a, b) Immunohistochemical detection of apoptotic hepatocytes using anti-ssDNA Abs in the livers from WT and IL-1ra KO mice at 2 h after APAP challenge. Representative results from six animals are shown in (a) (original magnification; × 200). The numbers of apoptotic hepatocytes per high-power microscopic field ( × 400) were enumerated and are shown in (b). All values represent means±s.e.m. (n=6 animals). *P<0.05, WT vs IL-1ra KO mice.

Leukocyte recruitment into the liver of WT and IL-1ra KO mice after APAP challenge. Immunohistochemical detection of neutrophils and macrophages in the livers was performed as described in ‘Materials and Methods’. Representative results on neutrophils (a, b) and macrophages (c, d) from six independent experiments are shown here (original magnification; × 200). The numbers of neutrophils (e) and macrophages (f) in the livers were determined at the indicated time intervals after APAP challenge (n=6). All values represent the mean±s.e.m. *P<0.05, **P<0.01, WT vs IL-1ra KO mice.

RT–PCR analysis for gene expression of IL-1α, IL-1β, CXCL1/KC, CXCL2/MIP-2, CCL2/MCP-1, and CCL3/MIP-1α in the livers of WT and IL-1ra KO mice at the indicated time intervals after APAP challenge. Representative results from six independent experiments are shown in (a). The ratios of IL-1α (b), IL-1β (c), CXCL1/KC (d), CXCL2/MIP-2 (e), CCL2/MCP-1 (f) and CCL3/MIP-1α (g) to β-actin in the livers of WT and IL-1ra KO mice are shown here. Each value represents mean±s.e.m. (n=6 animals). *P<0.05; **P<0.01, WT vs IL-1ra KO mice.
The Effects of Pretreatment with IL-1α or IL-1ra on APAP-Induced Liver Injury in WT Mice
Finally, we examined the effects of pretreatment of IL-1α on APAP-induced liver injury in vivo. When exogenous IL-1α was administered at 12 h before APAP challenge, it significantly diminished APAP-induced liver injury with reduced APAP adduct amounts (Figure 10a and b). Moreover, IL-1α reduced the intrahepatic expression levels of CYP1A2, CYP2E1, and CYP3A11 (Figure 10b). IL-1ra attenuated marginally APAP-induced increases in serum ALT levels when it was administered at 12 h before APAP challenge (data not shown), consistent with the previous report.17

The effects of IL-1α pretreatment on APAP-induced liver injury and intrahepatic expression of CYPs, and APAP adduct. WT mice were intraperitoneally administered recombinant IL-1α at 12 h before APAP challenge. (a) Analysis of serum ALT levels in IL-1α-treated WT (n=10) and PBS-treated WT (n=10) mice at 2, 6, 10, and 24 h after APAP challenge. All values represent the mean±s.e.m. *P<0.05; **P<0.01, IL-1α treatment vs PBS treatment. (b) Western blotting analysis of CYP1A2, CYP2E1, and CYP3A in the livers before and after APAP treatment. Representative results from six independent experiments are shown here.
DISCUSSION
Inflammatory cytokines, chemokines, and leukocytes are presumed to have detrimental roles in the pathogenesis of APAP-induced liver injury,5, 6, 7 whereas IL-6, IL-10, and IL-13 can protect the same liver injury.34, 35, 36 These observations would indicate the essential involvement of inflammatory responses in the development of liver damages caused by APAP.
Accumulating evidence implicates IL-1α and IL-1β as crucial mediators in various inflammatory reaction11, 13, 14, 15 and their endogenous antagonist, IL-1ra, can inhibit the biological activities of IL-1s by competitive binding to IL-1RI.8 We previously observed that the sequential treatment with killed Propioniobacteirum acnes and lipopolysaccharide (LPS) caused hepatic necrosis, along with enhanced intrahepatic expression of IL-1ra and IL-1s and that the neutralization of IL-1ra further augmented liver injury.18 This observation would indicate the protective role of IL-1ra in this liver injury model. Similarly, in APAP-induced liver injury, the intrahepatic expression of IL-1ra was enhanced together with IL-1s (unpublished data). Moreover, neutralization of IL-1 or administration of recombinant IL-1ra attenuated APAP-induced liver injury.16, 17 This led us to assume that endogenous IL-1ra can prevent or alleviate APAP-induced liver injury. Contrary to our hypothesis, we found that the genetic disruption of IL-1ra alleviated APAP hepatotoxicity.
Acetaminophen cannot directly cause hepatotoxicity. It is metabolized mainly by hepatocytes into NAPQI, which is detoxified by GSH. When generated NAPQI depletes GSH, NAPQI covalently binds to cysteine residues on intracellular proteins and induces the formation of 3-(cysteine-S-yl) APAP adducts in hepatocytes.37, 38 This process is the initial and irreversible step in the development of cell injury.38, 39 Among several adducts, selenium-binding protein with a molecular weight of 56 kDa, a major APAP adduct, reflects the amounts of generated NAPQI and APAP adducts,40, 41 and can determine the magnitude of APAP-induced direct liver injury.2, 3 IL-1ra KO-derived hepatocytes exhibited similar sensitivities to NAPQI as WT mouse-derived ones. Moreover, despite no differences of GSH levels, the intrahepatic amount of generated APAP adducts after APAP challenge, was less in IL-1ra KO mice than WT mice. Furthermore, blood APAP concentrations were significantly higher in IL-1ra KO mice than in WT mice after APAP challenge, implying that the absence of IL-1ra impaired APAP metabolism. Collectively, these observations would indicate that the reduced APAP-induced hepatotoxicity in IL-1ra KO mice can arise from decreased NAPQI generation from APAP.
Conversion of APAP into NAPQI is catalyzed by cytochrome P450 enzymes, particularly, CYP1A2, CYP2E1, and CYP3A11.26, 27, 28, 29, 30 Consistent with our assumption that IL-1ra KO mice may have a reduced capacity to generate NAPQI, the expression of these cytochrome P450 enzymes was diminished at both mRNA and protein levels in IL-1ra KO mice, as compared with WT mice. Proinflammatory cytokines such as IL-1, IL-6, and TNF-α can suppress CYP expression in hepatocytes.42, 43, 44, 45 Indeed, intrahepatic CYP expression was depressed in inflammation and infection, where these proinflammatory cytokines are presumed to be produced abundantly.32, 46 Similarly, Liu et al47 demonstrated that the pretreatment of LPS-sensitive C3H/OuJ mice but not LPS-resistant C3H/HeJ mice with a sublethal dose of LPS attenuated APAP hepatotoxicity by downregulating hepatic P450 enzymes in the livers. Moreover, the administration of both untreated C3H/OuJ and C3H/HeJ mice with IL-1α, alleviated APAP-induced liver injury.47 IL-1ra KO mice, which lack negative signals for IL-1, expressed IL-1α and IL-1β to a similar extent under unstimulated conditions, as WT mice. Thus, it is likely that IL-1-mediated signals were augmented in the livers of IL-1ra KO mice, compared with WT mice, thereby depressing intrahepatic CYP expression.
Several lines of evidence would indicate that the activation of nuclear factor-erythroid 2-related factor 2 (Nrf2) was essential for the detoxification of APAP by inducing the gene expression of phase 2 detoxifying enzymes such as glutathione S-transferases but not phase 1 enzymes such as cytochrome p450 enzymes.48, 49, 50 Thus, the differences of CYPs expression between WT and IL-1ra KO mice were not ascribable to Nrf2 activation. Another transcriptional factor, NF-κB, can be activated by various inflammatory stimuli including IL-1 and TNF-α,31 and is presumed to be involved in CYP gene transcription. Consistently, we demonstrated that IL-1α could enhance NF-κB activation and reciprocally suppressed the expression of CYP1A2, CYP2E1, and CYP3A in cultured WT-derived hepatocytes. APAP challenge augmented intrahepatic IL-1 expression and NF-κB activation to a larger extent in WT than IL-1ra KO mice but the intrahepatic expression of CYP1A2, CYP2E1, and CYP3A11 was still higher in WT mice than IL-1ra KO mice. This seemingly discrepancy may be explained by the fact that CYP expression was higher in untreated WT than IL-1ra KO mice. Higher CYP expression in untreated WT mice could not be cancelled out by APAP-induced IL-1 expression and NF-κB activation.
CYP1A1 and CYP1A2 gene transcription is under the control of aryl hydrocarbon receptor (AhR).51, 52 Once being activated, NF-κB can interact with AhR and subsequently prevent AhR from activating the transcription of the target genes including CYP1A1 and CYP1A2.53, 54 Sewer et al55 demonstrated that the intraperitoneal administration of LPS downregulated the gene expression of CYP2E1 in the livers of rats. Conversely, the administration of curcumin, a NF-κB inhibitor, reversed the gene transcription of CYP2E1 suppressed by LPS in the livers of rats, implying NF-κB as a negative regulator of CYP2E1 transcription.56 Pregnane X receptor (PXR), also known as steroid and xenobiotic receptor (SXR), can induce CYP3A11 gene transcription through dimerization with retinoid X receptor (RXR).57, 58 Activated NF-κB can form a heterodimer with RXR, thereby antagonizing PXR/SXR-mediated signaling.33, 59 These molecular mechanisms may account for downregulation of CYP genes by IL-142, 43, 44, 45 and suppressed CYP1A2, CYP2E1, and CYP3A11 expression observed in IL-1ra KO mice, which is presumed to exhibit enhanced IL-1-mediated signals.
Leukocyte infiltration starts following the appearance of tissue injury. Indeed, intrahepatic leukocyte infiltration becomes apparent later than 6 h after APAP challenge, when liver has already been injured directly by NAPQI toxicity and many hepatocytes became dead. The necrotic hepatocytes are a rich source of IL-1α,60 which can augment the expression of chemokines and adhesion molecules, thereby inducing the infiltration of leukocytes.61 Infiltrating leukocytes can produce injurious factors such as proinflammatory cytokines, reactive oxygen species, and NO.2, 3 The net result is the aggravation of the initial hepatocyte necrosis, which in turn augment IL-1α production and eventually leukocyte recruitment. APAP caused liver injury in mice lacking IFN-γ, TNF receptor p55, or CXC chemokine receptor 2, to similar extents as WT mice until 6 h after the challenge, but the injury was alleviated in these gene-deficient mice, thereafter.5, 6, 7 We further observed that these deficient mice expressed CYPs to similar extents as WT mice (our unpublished data) and therefore, it may be likely that these gene-deficient mice could generate similar amounts of NAPQI as WT mice. On the contrary, serum ALT levels and hepatocyte apoptosis were significantly attenuated in IL-1ra KO even at 2 h after APAP challenge. This may account for subsequent reduction in intrahepatic leukocyte recruitment and liver injury in IL-1ra KO mice, later than 6 h after APAP challenge. Moreover, reduced leukocyte recruitment diminished hepatocyte necrosis, and eventually attenuated IL-1 and chemokine expression in IL-1ra KO mice, because they were mainly produced by neutrophils and macrophages during APAP-induced liver injury. Thus, APAP-induced liver injury may consist of two phases: the initiation phase arises from direct hepatotoxicity caused by APAP-derived NAPQI and the later phase arises from leukocyte infiltration and leukocyte-derived cytokines in response to the liver injury in the initiation phase.
There are discrepancies on the effects of the IL-1 axis on APAP hepatotoxicity. Blazka et al17 demonstrated that IL-1ra reduced marginally APAP-induced liver injury, similarly as we observed here (our unpublished data). On the contrary, Liu et al47 demonstrated when IL-1α was given 12 or 24 h before APAP challenge, it attenuated susceptibility to APAP toxicity, together with reduced intrahepatic CYP expression, as similarly observed on IL-1ra KO mice. Thus, the IL-1 axis can suppress intrahepatic CYP expression with latency, thereby depressing NAPQI generation from APAP. Simultaneously, the IL-1 axis can perpetuate inflammatory reactions by inducing the expression of proinflammatory molecules such as TNF and chemokines in recruited leukocytes, thereby augmenting organ damage at the later phase. These double-edged actions of the IL-1 axis may account for the observed discrepancies.
The present observations indicate that the IL-1 axis has profound effects on APAP-induced toxicities. APAP is generally a safe antipyretic but can cause serious adverse effects to some patients.1 Given the presence of the polymorphisms of the IL-1 family genes,62 these polymorphisms of IL-1s may affect the sensitivities to APAP. If so, typing of human IL-1 family gene polymorphisms may help to decide the doses of APAP in individuals.
References
Lee WM, Acetaminophen and the U.S. Acute Liver Failure Study Group. Lowering the risks of hepatic failure. Hepatology 2004;40:6–9.
James LP, Mayeux PR, Hinson JA . Acetaminophen-induced hepatotoxicity. Drug Metab Dispos 2003;31:1499–1506.
Jaeschke H, Gores GJ, Cederbaum AI, et al. Mechanisms of hepatotoxicity. Toxicol Sci 2002;65:166–176.
Ishida Y, Kondo T, Kimura A, et al. Opposite roles of neutrophils and macrophages in the pathogenesis of acetaminophen-induced acute liver injury. Eur J Immunol 2006;36:1028–1038.
Ishida Y, Kondo T, Tsuneyama K, et al. The pathogenic roles of tumor necrosis factor receptor p55 in acetaminophen-induced liver injury in mice. J Leukoc Biol 2004;75:59–67.
Ishida Y, Kondo T, Ohshima T, et al. A pivotal involvement of IFN-γ in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J 2002;16:1227–1236.
Luster MI, Simeonova PP, Gallucci RM, et al. Role of inflammation in chemical-induced hepatotoxicity. Toxicol Lett 2001;120:317–321.
Dinarello CA . Interleukin-1. Cytokine Growth Factor Rev 1997;8: 253–265.
Eisenberg SP, Evans RJ, Arend WP, et al. Primary structure and functional expression from complementary DNA of a human interleukin-1 receptor antagonist. Nature 1990;343:341–346.
Hannum CH, Wilcox CJ, Arend WP, et al. Interleukin-1 receptor antagonist activity of a human interleukin-1 inhibitor. Nature 1990;343:336–340.
Arend WP, Malyak M, Guthridge CJ, et al. Interleukin-1 receptor antagonist: role in biology. Annu Rev Immunol 1998;16:27–55.
Dripps DJ, Brandhuber BJ, Thompson RC, et al. Interleukin-1 (IL-1) receptor antagonist binds to the 80-kDa IL-1 receptor but does not initiate IL-1 signal transduction. J Biol Chem 1991;266:10331–10336.
Fisher Jr CJ, Slotman GJ, Opal SM, et al. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit Care Med 1994;22:12–21.
Horai R, Saijo S, Tanioka H, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med 2000;191:313–320.
Ishida Y, Kondo T, Kimura A, et al. Absence of IL-1 receptor antagonist impaired wound healing along with aberrant NF-κB activation and a reciprocal suppression of TGF-β signal pathway. J Immunol 2006;176:5598–5606.
Blazka ME, Wilmer JL, Holladay SD, et al. Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol 1995;133:43–52.
Blazka ME, Elwell MR, Holladay SD, et al. Histopathology of acetaminophen-induced liver changes: role of interleukin 1 and tumor necrosis factor. Toxicol Pathol 1996;24:181–189.
Fujioka N, Mukaida N, Harada A, et al. Preparation of specific antibodies against murine IL-1ra and the establishment of IL-1ra as an endogenous regulator of bacteria-induced fulminant hepatitis in mice. J Leukoc Biol 1995;58:90–98.
Yasumoto K, Mukaida N, Harada A, et al. Molecular analysis of the cytokine network involved in cachexia in colon 26 adenocarcinoma-bearing mice. Cancer Res 1995;55:921–927.
Iizasa H, Yoneyama H, Mukaida N, et al. Exacerbation of granuloma formation in IL-1 receptor antagonist-deficient mice with impaired dendritic cell maturation associated with Th2 cytokine production. J Immunol 2005;174:3273–3280.
Moore M, Thor H, Moore G, et al. The toxicity of acetaminophen and N-acetyl-p-benzoquinone imine in isolated hepatocytes is associated with thiol depletion and increased cytosolic Ca2+. J Biol Chem 1985;260:13035–13040.
Hayashi T, Ishida Y, Kimura A, et al. IFN-γ protects cerulein-induced acute pancreatitis by repressing NF-κB activation. J Immunol 2007;178:7385–7394.
Bartholomew JB, Barge RB, Bolero SJ, et al. Purification, antibody production, and partial amino acid sequence of the 58-kDa acetaminophen-binding liver proteins. Toxicol Appl Pharmacol 1992;113:19–29.
Pumford NR, Martin BM, Hinson JA . A metabolite of acetaminophen covalently binds to the 56 kDa selenium binding protein. Biochem Biophys Res Commun 1992;182:1348–1355.
Baselt RC, Cravey RH . Disposition of Toxic Drugs and Chemicals in Man, 4th edn. Chemical Toxicology Institute: Foster City, CA, 1997, pp 2–5.
Tonge RP, Kelly EJ, Bruschi SA, et al. Role of CYP1A2 in the hepatotoxicity of acetaminophen: investigations using Cyp1a2 null mice. Toxicol Appl Pharmacol 1998;153:102–108.
Zaher H, Buters JT, Ward JM, et al. Protection against acetaminophen toxicity in CYP1A2 and CYP2E1 double-null mice. Toxicol Appl Pharmacol 1998;152:193–199.
Dai G, He L, Chou N, et al. Acetaminophen metabolism does not contribute to gender difference in its hepatotoxicity in mouse. Toxicol Sci 2006;92:33–41.
Dahlin DC, Miwa GT, Lu AY, et al. N-Acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci USA 1984;81:1327–1331.
Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993;6:511–518.
Cao Z, Tanaka M, Regnier C, et al. NF-κB activation by tumor necrosis factor and interleukin-1. Cold Spring Harb Symp Quant Biol 1999;64:473–483.
Riddick DS, Lee C, Bhathena A, et al. Transcriptional suppression of cytochrome P450 genes by endogenous and exogenous chemicals. Drug Metab Dispos 2004;32:367–375.
Zhou C, Tabb MM, Nelson EL, et al. Mutual repression between steroid and xenobiotic receptor and NF-κB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest 2006;116: 2280–2289.
Masubuchi Y, Bourdi M, Reilly TP, et al. Role of interleukin-6 in hepatic heat shock protein expression and protection against acetaminophen-induced liver disease. Biochem Biophys Res Commun 2003;304: 207–212.
Bourdi M, Masubuchi Y, Reilly TP, et al. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology 2002;35:289–298.
Yee SB, Bourdi M, Masson MJ, et al. Hepatoprotective role of endogenous interleukin-13 in a murine model of acetaminophen-induced liver disease. Chem Res Toxicol 2007;20:734–744.
Roberts DW, Pumford NR, Potter DW, et al. A sensitive immunochemical assay for acetaminophen-protein adducts. J Pharmacol Exp Ther 1987;241:527–533.
Pumford NR, Hinson JA, Potter DW, et al. Immunochemical quantitation of 3-(cystein-S-yl)acetaminophen adducts in serum and liver proteins of acetaminophen-treated mice. J Pharmacol Exp Ther 1989;248:190–196.
Gibson JD, Pumford NR, Samokyszyn VM, et al. Mechanism of acetaminophen-induced hepatotoxicity: covalent binding versus oxidative stress. Chem Res Toxicol 1996;9:580–585.
Cohen SD, Khairallah EA . Selective protein arylation and acetaminophen-induced hepatotoxicity. Drug Metab Rev 1997;29:59–77.
Hinson JA, Pumford NR, Roberts DW . Mechanisms of acetaminophen toxicity: immunochemical detection of drug-protein adducts. Drug Metab Rev 1995;27:73–92.
Barker CW, Fagan JB, Pasco DS . Interleukin-1β suppresses the induction of P4501A1 and P4501A2 mRNAs in isolated hepatocytes. J Biol Chem 1992;267:8050–8055.
Carlson TJ, Billings RE . Role of nitric oxide in the cytokine-mediated regulation of cytochrome P-450. Mol Pharmacol 1996;49:796–801.
Abdel-Razzak Z, Corcos L, Fautrel A, et al. Interleukin-1β antagonizes phenobarbital induction of several major cytochromes P450 in adult rat hepatocytes in primary culture. FEBS Lett 1995;366:159–164.
Abdel-Razzak Z, Loyer P, Fautrel A, et al. Cytokines down-regulate expression of major cytochrome P-450 enzymes in adult human hepatocytes in primary culture. Mol Pharmacol 1993;44:707–715.
Morgan ET . Regulation of cytochromes P450 during inflammation and infection. Drug Metab Rev 1997;29:1129–1188.
Liu J, Sendelbach LE, Parkinson A, et al. Endotoxin pretreatment protects against the hepatotoxicity of acetaminophen and carbon tetrachloride: role of cytochrome P450 suppression. Toxicology 2000;147:167–176.
Goldring CE, Kitteringham NR, Elsby R, et al. Activation of hepatic Nrf2 in vivo by acetaminophen in CD-1 mice. Hepatology 2004;39: 1267–1276.
LinksChan K, Han XD, Kan YW . An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci USA 2001;98:4611–4616.
Enomoto A, Itoh K, Nagayoshi E, et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci 2001;59:169–177.
Whitlock Jr JP . Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol 1999;39:103–125.
Ryu DY, Levi PE, Fernandez-Salguero P, et al. Piperonyl butoxide and acenaphthylene induce cytochrome P450 1A2 and 1B1 mRNA in aromatic hydrocarbon-responsive receptor knock-out mouse liver. Mol Pharmacol 1996;50:443–446.
Ke S, Rabson AB, Germino JF, et al. Mechanism of suppression of cytochrome P-450 1A1 expression by tumor necrosis factor-α and lipopolysaccharide. J Biol Chem 2001;276:39638–39644.
Tian Y, Ke S, Denison MS, et al. Ah receptor and NF-κB interactions, a potential mechanism for dioxin toxicity. J Biol Chem 1999;274:510–515.
Sewer MB, Koop DR, Morgan ET . Endotoxemia in rats is associated with induction of the P4504A subfamily and suppression of several other forms of cytochrome P450. Drug Metab Dispos 1996;24:401–407.
Cheng PY, Wang M, Morgan ET . Rapid transcriptional suppression of rat cytochrome P450 genes by endotoxin treatment and its inhibition by curcumin. J Pharmacol Exp Ther 2003;307:1205–1212.
Guo GL, Moffit JS, Nicol CJ, et al. Enhanced acetaminophen toxicity by activation of the pregnane X receptor. Toxicol Sci 2004;82:374–380.
Kliewer SA, Goodwin B, Willson TM . The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev 2002;23:687–702.
Gu X, Ke S, Liu D, et al. Role of NF-κB in regulation of PXR-mediated gene expression: a mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J Biol Chem 2006;281:17882–17889.
Sakurai T, He G, Matsuzawa A, et al. Hepatocyte necrosis induced by oxidative stress and IL-1α release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008;14:156–165.
Matsushima K, Oppenheim JJ . Interleukin 8 and MCAF: novel inflammatory cytokines inducible by IL-1 and TNF. Cytokine 1989;1:2–13.
Witkin SS, Gerber S, Ledger WJ . Influence of interleukin-1 receptor antagonist gene polymorphism on disease. Clin Infect Dis 2002;34:204–209.
Acknowledgements
We thank Ms Mariko Kawaguchi for her excellent assistant in the preparation of this paper. This study was financially supported in part by Grants-in-Aids for Scientific Research (B) and Young Scientists (B) from the Ministry of Education, Culture, Sports, Science, and Technology of the Japanese Government.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of Interest
No conflicts of interest were declared.
Rights and permissions
About this article
Cite this article
Ishibe, T., Kimura, A., Ishida, Y. et al. Reduced acetaminophen-induced liver injury in mice by genetic disruption of IL-1 receptor antagonist. Lab Invest 89, 68–79 (2009). https://doi.org/10.1038/labinvest.2008.110
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2008.110
Keywords
This article is cited by
-
Involvement of hepatic stellate cell cytoglobin in acute hepatocyte damage through the regulation of CYP2E1-mediated xenobiotic metabolism
Laboratory Investigation (2015)
-
Application of IL-36 receptor antagonist weakens CCL20 expression and impairs recovery in the late phase of murine acetaminophen-induced liver injury
Scientific Reports (2015)
-
The dual role of osteopontin in acetaminophen hepatotoxicity
Acta Pharmacologica Sinica (2012)
-
rhIL-1Ra reduces hepatocellular apoptosis in mice with acetaminophen-induced acute liver failure
Laboratory Investigation (2010)
-
The potential of cytokines as safety biomarkers for drug-induced liver injury
European Journal of Clinical Pharmacology (2010)
