
Abstract
Hutchinson–Gilford progeria syndrome (HGPS) is an extremely rare genetic disorder that shows a characteristic progeria phenotype. We conducted a questionnaire survey of 1173 tertiary hospitals in Japan and reviewed the academic reports, to identify the characteristics of Asian patients with classical HGPS. As a result, four Japanese patients were identified; this was estimated to account for approximately two-third of the prevalence in Japan. Three Asian patients who had definitively been diagnosed with classical HGPS were identified in the literature; in total, the clinical characteristics of seven patients were evaluated. Most of the clinical phenotypes of Asian patients were essentially similar to those of patients of other ethnicities, such as sclerodermatous skin, growth failure, loss of scalp hair or severe complications of cardiovascular and cerebral ischemic disease. In conclusion, to circumvent or minimalize severe vascular complication, an early diagnosis, careful observation and, promisingly, new intervention with farnesylation inhibitors may improve the prognosis of classical HGPS patients.
Similar content being viewed by others
Introduction
Hutchinson–Gilford progeria syndrome (HGPS) is a rare genetic disorder that shows a relatively severe phenotype among the hereditary progeria syndromes.1 According to a report from the Progeria Research Foundation, which was published on 1 April 2017, 148 patients have been reported (https://www.progeriaresearch.org/) and it was estimated that there were ~350 to 400 children living with HGPS worldwide. The typical symptoms, such as a disproportionally large head for face, loss of scalp hair, a small chin and sclerodermatous skin gradually progress from ~6 months of age; in contrast, the patient’s psychomotor development is preserved and remains almost within the normal ranges.1, 2, 3, 4 In the later years of life, typically in their early teens, classical-type HGPS patients develop severe, prominent multisystem complications, such as cerebral infarction, coronary artery disease, cardiac valvulopathy and hypertension.1, 2, 3 The average life expectancy of HGPS patients is reported to be ~14.6 years.1 In recent years, treatment with farnesylation inhibitors of progerin has been expected to improve the clinical symptoms, 5, 6, 7, 8 and the new therapies currently under human clinical trial investigation for HGPS include lonafarnib plus everolimus (https://clinicaltrials.gov/). Thus, an early diagnosis and intervention are critically important.
Both phenotype and genetic confirmation of a LMNA mutation are essential for the diagnosis of HGPS; classic type is defined based on the recognition of common clinical features and the presence of the c.1824C>T (p.Gly608Gly) heterozygous pathogenic variant in the LMNA gene.9, 10 The diagnosis of the atypical type is made in individuals with clinical features that are similar to classic HGPS, who have progerin-producing pathogenic variants in intron 11 of LMNA, including c.1822G>A, c.1968+1G>A, c.1968+2 T>A, c.1968+2 T>C and c.1968+5G>C.1, 11
Many Asian patients with progeria or progeria-like phenotypes have been reported in academic papers; however, genetic studies were not always carried out. According to the report from the Progeria Research Foundation, 246 children were registered and 32% of them (N=79) were Asian; their phenotype was not reported on the Progeria Research Foundation website (https://www.progeriaresearch.org/prf-by-the-numbers/). To evaluate the phenotypes in Asian patients with classical HGPS, we conducted a nationwide questionnaire survey in Japan and reviewed the academic reports on classical HGPS in Asian countries to discuss whether any of the characteristic phenotypes were characteristically found in Asian patients.
Materials and methods
The methods of the questionnaire survey
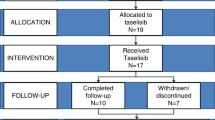
We conducted a primary survey by mailing a questionnaire to the pediatric departments of 1173 hospitals that contained more than 200 beds for in-patients. We first asked the pediatricians at the hospitals whether they had experienced HGPS patients between 2008 and 2013. Next, we carried out a secondary survey on the clinical characteristics including the physical findings and complications of the HGPS candidates. Finally, we conducted a third survey and obtained detailed clinical information from eight patients (Figure 1).

Study flow diagram of the screening or literature survey for HGPS patients in Japan.
Ethical considerations
The present study was conducted in compliance with the Declaration of Helsinki. The information of the patients was managed with consideration of their privacy. When a patient or the patient’s family did not consent to their inclusion in the study, the patient was excluded from the study. The study was reviewed and approved by the research ethics committee of the Faculty of Medicine in Oita University (Approval number: 919).
Literature review
We searched PubMed, Web of Science and EMBASE, for all of the reported cases of ‘HGPS’ in Asian countries, including India, Indonesia, Cambodia, Singapore, Sri Lanka, Thailand, Korea, China, Nepal, Pakistan, Bangladesh, East Timor, Philippines, Bhutan, Brunei, Vietnam, Malaysia, Myanmar, the Maldives, Mongolia and Laos, as well as Japan.
Results
The patients from the Japanese national questionnaire survey
Fifteen candidates with probable HGPS were identified in the primary screening. The patient information from the questionnaires was carefully screened and some of these patients were considered to be non-HGPS patients and were excluded from further study. Ten candidate hospitals completed a secondary questionnaire survey. We obtained the general clinical information of nine patients with HGPS from the secondary questionnaires. We also investigated the patient reports from academic conferences in Japan, contacted the authors/speakers by e-mail or telephone and finally confirmed one additional Japanese patient. The two survey methods identified 10 patients. One patient who did not give their consent and one patient who did not respond to the third questionnaire were excluded from the subsequent analysis. Consequently, the detailed clinical information of eight Japanese patients (male, n=4; female, n=4) were collected. The mutation specific for classical HGPS, c.1824C>T in the LMNA gene, was identified in four of these patients.
The patients from the literature review
From the search in May 2017, 30 Asian cases with HGPS were identified; however, few of them were genetically analyzed and only 3 had the c.1824C>T mutation in the LMNA gene; 2 were Chinese12, 13 and 1 was Korean.14
The clinical characteristics of Asian patients with classical HGPS
The detailed clinical information of classical HGPS from four Japanese, two Chinese and one Korean patients (male, n=4; female, n=3) are summarized in Table 1.
Growth characteristics
To identify the growth characteristics of classical HGPS patients, we focused on the patients whose physical data were closely recorded and evaluated on the growth charts for Japanese boys or girls. Growth failure appeared immediately after birth in all Japanese cases. In three of them, the body weight was below the third percentile within the first 6 months of life and the height was below the third percentile within 1 year after birth. In the remaining one case, the height and weight were below the third percentile by 3.5 years of age. As for the two other Asian cases, the body size (in terms of height and weight) of the patients, at the time of reporting, was below the third percentile. A pubertal growth spurt with marked weight gain was not observed in the two patients (Case 3 and 4) who were evaluated when they were older than 12 years of age (Figure 2).

Growth curves summarizing growth data of four Japanese patients. Filled circles, squares or triangles represent the heights or weights of boys (left side in black) and girls (right side in red). A full color version of this figure is available at the Journal of Human Genetics journal online.
Body appearance
The characteristic body appearance, caused by the reduction of subcutaneous fat was recognized. Sclerodermatous skin and loss of scalp hair were observed in all patients (seven out of seven), especially the sclerodermatous skin, which appeared as early as 2 months after birth, indicating that this finding would be an important clinical clue for an early diagnosis during infancy. In contrast, the timing of the loss of scalp hair was also commonly observed and it seemed to be variable during their early stage of life. This was probably dependent on the severity of growth failure in their early postnatal period.
Oral/dental findings
Among the typical findings regarding the dental appearance of HGPS patients, information regarding the delayed eruption of the primary teeth was obtained in onecase.
Skeletal system/joints
Micrognathia and osteoarthritis were observed in all four Japanese patients. Joint contracture and a decreased range of joint motion were not fully evaluated and are not shown in the Table 1.
Cardiovascular/neurovascular complications
Cerebrovascular or cardiovascular complications due to severe arteriosclerosis occurred in three of three Japanese patients who were evaluated at between 10 and 15 years of age. Dyslipidemia, hypertension and diabetes were not distinctive in the evaluated patients.
Discussion
HGPS (OMIM 176670) was first reported by Hutchinson15 and was independently reported by Gilford.16 Based on previous papers, the prevalence is estimated to be ~1 in 18 million.1 Based on this prevalence and the Japanese population of ~120 million (in 2017), it is estimated that there are 6 HGPS patients in Japan. To understand the clinical characteristics of this extremely rare genetic disease, we first carried out a nationwide survey to identify the Japanese patients with classical type HGPS. As a result, four patients were identified; this accounts for two-third of the estimated prevalence. Three other Asian patients who had been reported in the literature and who had received a definite diagnosis of classical HGPS were also included. Thus, the clinical characteristics of seven patients were evaluated.
In this study we identified several findings that are worth mentioning. First, most of the clinical phenotypes in Asian patients were essentially similar to those in other ethnicities, such as sclerodermatou skin, growth failure, loss of scalp hair or severe complications of cardiovascular and cerebrovascular ischemic disease. Severe growth impairment and sclerodermatous skin appeared in very early infancy, the same as in previous reports.4, 17 These symptoms may be essential to making an early diagnosis of classical HGPS in the Asian population as well. Second, the incidence of cerebral hemorrhage or infarction seemed higher in comparison with previous reports; all (three out of three) of the Japanese patients who were older than 10 years of age suffered from cerebral infarction and the two affected children died from cerebral hemorrhage or infarction, whereas the affected children died from cardiovascular disease in the previous reports.18, 19 The numbers were too small to conclude this finding to be significant; however, it might reflect a tendency in Asian patients. Finally, several family members or their attending doctors obstinately refused to answer the questionnaires. They told us that they had long suffered from being looked at with a great deal of curiosity from their community and the mass media. The medical experts should take this serious situation into consideration to the utmost extent and we must carefully promote educational or enlightenment activities for HGPS patients, to facilitate the early diagnosis and treatment.
In conclusion, the survey of HGPS patients in Asian population demonstrated that the characteristic symptoms were essentially the same as those shown in the literature from other countries. To avoid the severe vascular complications associated with severe HGPS, especially cerebrovascular hemorrhage and ischemia, an early diagnosis and careful observation are important. The genetic examination of the LMNA gene is essential for the diagnosis of classical or other types of progeria; thus, the LMNA gene should be analyzed with careful consideration in children who display the characteristic phenotypes and in whom HGPS is suspected.
References
Gordon, L. B., Brown, W. T., Collins, F. S. in GeneReviews(R)(eds Pagon, R. A., Adam, M. P., Ardinger, H. H., Wallace, S. E., Amemiya, A., Bean, L. J. H. et al (1993). https://www.ncbi.nlm.nih.gov/pubmed/20301300.
Hennekam, R. C. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am. J. Med. Genet. A 140, 2603–2624 (2006).
Merideth, M. A., Gordon, L. B., Clauss, S., Sachdev, V., Smith, A. C., Perry, M. B. et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 358, 592–604 (2008).
Mazereeuw-Hautier, J., Wilson, L. C., Mohammed, S., Smallwood, D., Shackleton, S., Atherton, D. J. et al. Hutchinson-Gilford progeria syndrome: clinical findings in three patients carrying the G608G mutation in LMNA and review of the literature. Br. J. Dermatol. 156, 1308–1314 (2007).
Gordon, L. B., Kleinman, M. E., Massaro, J., D'Agostino, R. B. Sr, Shappell, H., Gerhard-Herman, M. et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation 134, 114–125 (2016).
Gordon, L. B., Massaro, J., D'Agostino, R. B. Sr, Campbell, S. E., Brazier, J., Brown, W. T. et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 130, 27–34 (2014).
McClintock, D., Gordon, L. B. & Djabali, K. Hutchinson-Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proc. Natl Acad. Sci. USA 103, 2154–2159 (2006).
Young, S. G., Meta, M., Yang, S. H. & Fong, L. G. Prelamin A farnesylation and progeroid syndromes. J. Biol. Chem. 281, 39741–39745 (2006).
Eriksson, M., Brown, W. T., Gordon, L. B., Glynn, M. W., Singer, J., Scott, L. et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298 (2003).
De Sandre-Giovannoli, A., Bernard, R., Cau, P., Navarro, C., Amiel, J., Boccaccio, I. et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 300, 2055 (2003).
Moulson, C. L., Fong, L. G., Gardner, J. M., Farber, E. A., Go, G., Passariello, A. et al. Increased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromes. Hum. Mutat. 28, 882–889 (2007).
Chu, Y., Xu, Z. G., Xu, Z. & Ma, L. Hutchinson-Gilford progeria syndrome caused by an LMNA mutation: a case report. Pediatr. Dermatol. 32, 271–275 (2015).
Zhang, H., Chen, X., Guo, Y., Liang, J., Tang, L., Yu, H. et al. Hutchinson-Gilford progeria syndrome: report of 2 cases and a novel LMNA mutation of HGPS in China. J. Am. Acad. Dermatol. 69, e175–e176 (2013).
Kim, H. K., Lee, J. Y., Bae, E. J., Oh, P. S., Park, W. I., Lee, D. S. et al. Hutchinson-Gilford progeria syndrome with G608G LMNA mutation. J. Korean Med. Sci. 26, 1642–1645 (2011).
Hutchinson, J. Congenital absence of hair and mammary glands with atrophic condition of the skin and its appendages, in a boy whose mother had been almost wholly bald from alopecia areata from the age of six. Med. Chir. Trans. 69, 473–477 (1886).
Gilford, H. Progeria: a form of senilism. Practitioner 73, 188–217 (1904).
Rork, J. F., Huang, J. T., Gordon, L. B., Kleinman, M., Kieran, M. W. & Liang, M. G. Initial cutaneous manifestations of Hutchinson-Gilford progeria syndrome. Pediatr. Dermatol. 31, 196–202 (2014).
Silvera, V. M., Gordon, L. B., Orbach, D. B., Campbell, S. E., Machan, J. T. & Ullrich, N. J. Imaging characteristics of cerebrovascular arteriopathy and stroke in Hutchinson-Gilford progeria syndrome. AJNR Am. J. Neuroradiol. 34, 1091–1097 (2013).
Gerhard-Herman, M., Smoot, L. B., Wake, N., Kieran, M. W., Kleinman, M. E., Miller, D. T. et al. Mechanisms of premature vascular aging in children with Hutchinson-Gilford progeria syndrome. Hypertension 59, 92–97 (2012).
Acknowledgements
We thank Dr Brian Quinn for his support and writing assistance, and Dr Hiroaki Miyahara for his help in making the figures. This work was supported by Health, Labour and Welfare Sciences Research Grants that were provided by the Ministry of Health, Labour and Welfare for research on rare and by the Acceleration Program for Intractable Diseases Research utilizing disease-specific iPS cells from Japan Agency for Medical Research and Development, AMED. We thank all patients and their family for supporting our study. We thank Dr Eiko Kato (Department of Pediatrics, Tosei General Hospital, Seto), Dr Tadataka Hoshika (Department of Pediatrics, Tottori Prefectural Central Hospital, Tottori), Dr Yasuhiko Takahashi (Department of Pediatrics, JCHO Kyushu Hospital, Kitakyushu), Dr Hiroshi Mochizuki (Department of Endocrinology and Metabolism, Saitama Children's Medical Center, Saitama), Dr Katsuhiro Hayashi (Department of Orthopaedic Surgery, Kanazawa University Hospital, Kanazawa) and Dr Naohiro Suga (Department of Pediatrics, Tagawa Municipal Hospital, Tagawa) for providing clinical information of the patients.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Sato-Kawano, N., Takemoto, M., Okabe, E. et al. The clinical characteristics of Asian patients with classical-type Hutchinson–Gilford progeria syndrome. J Hum Genet 62, 1031–1035 (2017). https://doi.org/10.1038/jhg.2017.90
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.90
