
Abstract
Split hand/foot malformation (SHFM) is a congenital limb deficiency with missing or shortened central digits. Some SHFM genes have been identified but the cause of many SHFM cases is unknown. We used single-nucleotide polymorphism (SNP) microarray analysis to detect copy-number variants (CNVs) in 25 SHFM cases without other birth defects from New York State (NYS), prioritized CNVs absent from population CNV databases, and validated these CNVs using quantitative real-time polymerase chain reaction (qPCR). We tested for the validated CNVs in seven cases from Iowa using qPCR, and also sequenced 36 SHFM candidate genes in all the subjects. Seven NYS cases had a potentially deleterious variant: two had a p.R225H or p.R225L mutation in TP63, one had a 17q25 microdeletion, one had a 10q24 microduplication and three had a 17p13.3 microduplication. In addition, one Iowa case had a de novo 10q24 microduplication. The 17q25 microdeletion has not been reported previously in SHFM and included two SHFM candidate genes (SUMO2 and GRB2), while the 10q24 and 17p13.3 CNVs had breakpoints within genomic regions that contained putative regulatory elements and a limb development gene. In SHFM pathogenesis, the microdeletion may cause haploinsufficiency of SHFM genes and/or deletion of their regulatory regions, and the microduplications could disrupt regulatory elements that control transcription of limb development genes.
Similar content being viewed by others

Introduction
Split hand/foot malformation (SHFM) is a distal limb malformation involving missing or shortened central digits, often in association with fusion of the remaining digits and median clefts of the hands and/or feet.1 SHFM has an estimated prevalence of 1–9 per 100 000 births,2, 3, 4 represents 1–15% of congenital limb deficiencies,2, 3, 4 occurs in nonsyndromic and syndromic forms5 and displays phenotypic variability even within families.6 Loss of the central portion of the apical ectodermal ridge (AER), a critical signaling center for distal limb outgrowth and digit development located at the apex of the limb bud,7 is thought to be responsible for SHFM.8 In humans, the AER is visible in the developing upper limb by embryonic day (E) 32 after conception (gestational week 7).9, 10 The bones and musculature of the human limb are established by E56 (gestational week 10);10 therefore, SHFM likely occurs during the seventh to ninth week of gestation.
Multiple genetic loci have been associated with SHFM, including mutations in TP63,5 WNT10B,6 CDH3,11 DLX5,12 FGFR1,13 FGFR214 and MAP3K20.15 Reported copy-number gains at chromosome 10q2416 and 17p13.3,17 microdeletions at chromosome 2q31,18 chromosome rearrangements at 2q1419 and 6q21,20 and linkage to Xq2621 in SHFM patients suggest that more SHFM genes, as yet undetermined, are present at these and other loci. Because our knowledge of the genetic causes of SHFM is incomplete, we scanned genome-wide for copy-number variants (CNVs) and performed targeted sequencing of candidate genes to search for genetic variants involved in SHFM.
Materials and methods
Subjects
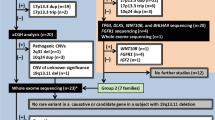
New York State (NYS) has mandatory reporting of major structural birth defects identified within the first 2 years of life to the NYS Congenital Malformations Registry.22 Each birth defect is coded using the expanded British Paediatric Association coding system based on hospital-provided descriptions entered as a text field and reviewed by a clinician as needed. We searched the Congenital Malformations Registry for isolated SHFM cases, here defined as subjects who had SHFM without additional major structural birth defects. We queried Congenital Malformations Registry records using the British Paediatric Association codes corresponding to congenital absence of fingers (755.247), congenital cleft hand (755.250), absence of preaxial fingers (755.2609), absence of postaxial fingers (755.2709), congenital absence of foot or toes (755.3401), congenital absence of toe (755.3409), congenital cleft foot (755.350) and absent digits—not otherwise specified (755.440). We selected cases that had ectrodactyly, cleft hand and/or foot, or absent central fingers/toes/digits/phalanges mentioned in the narrative description of the limb defect in the Congenital Malformations Registry record and that did not have the British Paediatric Association codes indicating the presence of other major birth defects or chromosomal anomalies. In total, 25 isolated SHFM cases were identified from all live births occurring in NYS from 1998 to 2005 (n=2 023 049). We also selected five controls with no known major birth defects from among NYS live births delivered during the same time period to use as technical controls for microarray genotyping.
We used single-nucleotide polymorphism (SNP) microarrays to detect CNVs and quantitative real-time polymerase chain reaction (qPCR) assays to validate CNVs in NYS cases and controls, and used another group of SHFM cases and controls from Iowa to test for CNVs that we validated in NYS SHFM cases. Seven isolated SHFM cases and seven controls without major birth defects, and their parents, were available from among live births delivered from 1999 to 2009 in Iowa. The medical records of the Iowa cases were reviewed by clinical geneticists to confirm the diagnosis of SHFM and the absence of other major birth defects. Iowa subjects were examined for validated CNVs using qPCR assays. All NYS and Iowa cases and controls were included in targeted sequencing assays of SHFM candidate genes.
The NYS Department of Health Institutional Review Board, the University of Iowa Institutional Review Board and the National Institutes of Health—Office of Human Subjects Research Protections approved this study.
DNA specimens
For each NYS case and control, DNA was obtained from residual blood spots archived by the NYS Newborn Screening Program. The DNA was extracted from two 3-mm dried blood spot punches using a laboratory-developed method.23 For Iowa case– and control–parent trios, DNA that had been extracted from buccal swabs due to the families’ participation in the National Birth Defects Prevention Study24 was used.
Genotyping
DNA specimens from NYS cases and controls were used for microarray genotyping. Genotyping was performed at the Johns Hopkins University SNP Center using the HumanOmni2.5–4 array and the Infinium HD assay protocol (Illumina, San Diego, CA, USA). One control specimen and one case specimen were run in duplicate and served as quality control specimens. Data were analyzed using Illumina GenomeStudio version 2011.1. The genotype no-call threshold was set at <0.15. Genotypes were called using genotype clusters defined based on (i) the standard cluster file provided by Illumina and (ii) the data generated in this project. For each of the two sets of genotype calls, genotypes were manually reviewed, re-clustered, edited and excluded (where appropriate) based on parameters and quality control metrics described in Illumina’s Infinium Genotyping Data Analysis Technical Note (http://res.illumina.com/documents/products/technotes/technote_infinium_genotyping_data_analysis.pdf).
CNV calling and annotation
CNVs were imputed from both sets of SNP genotype data (standard cluster file and custom cluster file) using Illumina’s cnvPartition algorithm (version 3.1.6) and the PennCNV algorithm.25 Each CNV call required a threshold of three SNP probes. For both cnvPartition and PennCNV, the data were GC wave-adjusted to reduce false positive calls. Default confidence values were used: 35 for cnvPartition and 10 for PennCNV. CNV call files were compiled and annotated, and the percent overlap with each of the following control databases was assessed: HapMap common CNVs,26 Children’s Hospital of Philadelphia (CHOP) database of CNVs in healthy individuals27 and the Database of Genomic Variants (DGV).28 The following were also noted: the percent agreement between cnvPartition calls and PennCNV calls, the number of cases and controls with the same/overlapping CNVs, and the transcripts and genes encompassed by each CNV. Transcripts and genes were identified using GENCODE Genes track (version 12, HAVANA and Ensembl Datasets).29 CNV calls were reviewed for overlap with genes in the Online Mendelian Inheritance in Man database,30 pathogenic CNVs defined by the Internal Standards for Cytogenomic Arrays database31 (accessed via the University of California—Santa Cruz (UCSC) Genome Browser32), CNVs previously reported in SHFM cases, genes associated with SHFM, and variants in the DECIPHER database33 associated with phenotype descriptions that included SHFM.
CNV validation
CNVs were selected for validation if they (i) had been detected in SHFM, either in previous reports or the DECIPHER database, (ii) overlapped genes mutated in SHFM or (iii) overlapped the coding region of at least one gene in one or more NYS SHFM cases, were not present in NYS controls and contained at least one gene that was not overlapped by CNVs reported in population databases of CNVs (HapMap, CHOP database, and DGV). The procedure for validation by qPCR is detailed in the Supplementary Methods.
Targeted sequencing of SHFM candidate genes
Forty-nine candidate genes were selected for sequencing because mutations in these genes have been detected in SFHM cases or other patients with congenitally missing digits, the genes are located in or near CNVs or other chromosomal rearrangements detected in cases in this study or in previous reports, or disruption of the genes results in a reduced number of digits in animal models. Only the coding regions and exon–intron boundaries of the genes were sequenced. Targeted sequencing of DNA specimens from NYS and Iowa cases and controls was performed using a custom Ion AmpliSeq panel (ThermoFisher Scientific, Waltham, MA, USA) and sequence variants were annotated using the ANNOVAR34 program. The details of sequencing and variant annotation are provided in Supplementary Methods.
Selection of potentially pathogenic sequence variants
Thirteen of 49 genes targeted for sequencing were in CNVs that failed to validate by qPCR in our study subjects, and we eliminated these genes from further analysis. Our analysis used the data from the remaining 36 genes (Supplementary Table S1). Annotated variants were filtered to select only those that met all of the following criteria: a quality score ⩾20, a flow evaluator alternate allele observation count ⩾20, an allele frequency in any reference population (obtained from ANNOVAR’s popfreq_all_20150413 database, which contains allele frequencies compiled from several population databases) <0.01, absent from control samples, located in exons or canonical splice sites, and variants were nonsynonymous, nonsense, frameshift or in-frame insertions/deletions. An additional, nonsynonymous TP63 variant (p.P417T) that had a minor allele frequency of 0.011 in the Complete Genomics 46 (CG46) database (used by ANNOVAR for annotation) was also selected because TP63 is a known SHFM gene. We considered that TP63 p.P417T has a minor allele frequency of 0.0029 in the ExAC database,35 and the CG46 database’s small sample size could be responsible for its higher TP63 p.P417T minor allele frequency.
Validation of selected sequence variants
Sanger sequencing was used to validate the selected sequence variants. The sequencing procedure is described in the Supplementary Methods and the conditions used for PCR are provided in Supplementary Table S2.
Statistical analysis
We used NYS birth certificates to obtain data on maternal age at delivery, race/ethnicity, education at delivery, parity and smoking during pregnancy, as well as infant sex for NYS SHFM cases and all NYS live births delivered from 1998 to 2005. We compared the data between these groups using the chi-squared test, and considered P<0.05 to represent statistical significance.
Bioinformatics analysis
We accessed transcriptome profiling (RNA-seq) data and chromatin immunoprecipitation (ChIP-seq) data on the acetylation of lysine 27 of the H3 histone protein (H3K27ac) for mouse and human limb buds through the National Center for Biotechnology Information—Gene Expression Omnibus (GEO) data repository. The data were generated by Cotney et al.36 and DeMare et al.,37 and had GEO accession numbers GSE42237 and GSE42413. The data were viewed using the UCSC Genome Browser.
Results
The estimated prevalence of isolated SHFM cases without other major birth defects in NYS from 1998 to 2005 was 1.24 per 100 000 (25/2 023 049) live births. Maternal age at delivery, race/ethnicity, education at delivery, parity, smoking during pregnancy and infant sex did not differ significantly different between NYS cases and all NYS live births during this time period (Supplementary Table S3).
In NYS cases, we observed 30 CNVs that had been reported previously in SHFM or that overlapped the coding region of at least one gene and were absent from NYS controls and from population databases of CNVs. Heterozygous CNVs at three regions (Table 1) were validated by qPCR (targeted genomic loci used for validation are listed in Supplementary Table S4). It is possible that the other 27 CNVs did not validate because of their small size. The median (interquartile range) size for the three validated compared with the 27 nonvalidated CNVs was 175.5 (168.7–306.6) kb vs 29.9 (14.5–55.0) kb, respectively (P=0.011; Wilcoxon rank-sum test). Most of the 27 CNVs were probably too small to be reliably imputed from the genotype data, especially because there was noise in clustering genotypes on a small number of samples (25 cases and five controls). Despite their small size, we attempted to validate the 27 CNVs because we did not want to overlook a CNV that intersected a potentially novel SHFM gene.
Five NYS cases had a validated CNV. One case had a 10q24 duplication (Supplementary Figure S1), another three cases each had a 17p13.3 duplication (Supplementary Figure S2) and an additional case had a 17q25 deletion (Supplementary Figure S3) not described previously in SHFM; the 17q25 CNV region contained 11 genes (Supplementary Table S5). With the use of qPCR, we tested for CNVs at the 10q24, 17p13.3 and 17q25 loci in Iowa cases and controls. One of the Iowa cases and none of the Iowa controls had a 10q24 duplication that overlapped the genomic location of the 10q24 CNV in the NYS case (Supplementary Table S4). Evaluation by qPCR detected no CNVs at 10q24 in the parents of the Iowa case, indicating that the duplication was de novo. The analysis of three polymorphic microsatellite markers in this family did not demonstrate inconsistency with Mendelian inheritance. None of the Iowa cases and controls had CNVs in the 17p13.3 or 17q25 region.
To determine whether our SHFM cases had other point mutations that might cause SHFM, we performed targeted next-generation sequencing of 36 SHFM candidate genes (listed in Supplementary Table S1) in NYS and Iowa cases and controls. In cases, we prioritized 38 sequence variants that met our criteria for potential pathogenicity. Fourteen variants, all nonsynonymous and heterozygous, were validated by Sanger sequencing (Table 2; Supplementary Figure S4). Two NYS cases had private variants at amino acid R225 of TP63; R225 is in the DNA-binding domain of TP63 and is highly conserved based on multiple alignment of the TP63 protein from 10 vertebrates (Supplementary Figure 5a). Eight mutation predictor tools were used to evaluate the 14 validated variants for potential functional impact, and all predicted that the two R225 variants would be damaging (Table 2; Supplementary Table S6). Another validated variant in TP63, p.P417T, also modifies a conserved residue (Supplementary Figure 5b) and is located in a proline-rich region between the tetramerization and sterile alpha motif domains (based on protein domains reported for the TP63 protein with GenBank accession number NP_001108452). Seven mutation predictor tools predicted a deleterious effect of this variant but one tool predicted low functional impact (Supplementary Table S6). For the other 11 variants, the number of tools predicting a deleterious effect ranged from one to five (Table 2; Supplementary Table S6). Because mutation predictor tools lacked consensus on the potential pathogenicity of the latter 12 variants, we did not focus on these variants as potential causes of SHFM.
To delineate the critical regions of the 17q25, 10q24 and 17p13.3 CNVs in SHFM, we compared the genomic coordinates of the validated CNVs from this study with those of previous SHFM reports and DECIPHER database cases that had a limb phenotype description consistent with SHFM. The 17q25 copy-number loss in patient 16 partially overlapped a microdeletion in a patient from the DECIPHER database (Figure 1). The overlap extended from the second intron of ARMC7 to the seventh intron of NUP85. Chromosome 10q24 CNVs were copy-number gains that, except for two CNVs, had at least one breakpoint within the region that extended from LBX1 to FGF8 (Figure 2; genomic coordinates and references listed in Supplementary Table S7). Chromosome 17p13.3 CNVs were copy-number gains (with one exception) that either overlapped the region extending from ABR to the intergenic region immediately downstream of TUSC5 or had a breakpoint within this region (Figure 3; genomic coordinates and references provided in Supplementary Table S8). Genes in all three CNV regions are expressed in developing human limb at E44 (Supplementary Figures S6–S8).

Region of copy-number loss at chromosome 17q25 in patients with split hand/foot malformation (GRCh37/hg19 assembly). The DECIPHER case was reported with a microdeletion at chr17:72952528-73214654 and the following phenotypes: aplasia of the fingers, cutaneous finger syndactyly, asymmetry of the mandible and hip dislocation. A full color version of this figure is available at the Journal of Human Genetics journal online.

Region of copy-number variation at chromosome 10q24 among patients with split hand/foot malformation (GRCh37/hg19 assembly). All variants were copy-number gains. Breakpoints and references to patient reports are listed in Supplementary Table S7. A full color version of this figure is available at the Journal of Human Genetics journal online.

The overlap of copy-number variation at chromosome 17p13.3 in patients with split hand/foot malformation (GRCh37/hg19 assembly). The DECIPHER 282751 case had a copy-number loss; all other cases had a copy-number gain. Copy-number variant breakpoints and references to patient reports are listed in Supplementary Table S8. A full color version of this figure is available at the Journal of Human Genetics journal online.
The evolutionarily conserved gene order of the Lbx1-Fgf8 region led to the recognition that this region contains a group of predicted enhancers, arranged in a precise spatial pattern, that coordinately control Fgf8 expression during embryonic development.38, 39, 40 Chromosomal rearrangements of this region engineered in mouse embryos suggested that copy-number gains at 10q24 might cause SHFM by disrupting the spatial arrangement of the enhancers leading to dysregulation of gene expression.40 In human limb buds, the subgroup of predicted enhancers shown to drive reproducible patterns of reporter gene expression in transgenic mouse embryos (Supplementary Table S9) coincided with peaks of histone H3K27ac modifications, often found near active enhancers41 (Supplementary Figure S9). This provides support for the involvement of predicted enhancers at LBX1-FGF8 in regulating gene expression during limb development. Because chromosome 17p13.3 copy-number gains are also associated with SHFM, we hypothesized that these CNVs could be disrupting regulatory elements in the 17p13.3 region. Gene order in the ABR-TUSC5 region is conserved among vertebrates (Supplementary Figure S10), implying that if regulatory elements are located in this region, their spatial orientation could be relevant to their regulation of gene expression. In human and mouse limb buds, there were peaks of histone H3K27ac modifications in conserved, noncoding regions at ABR-TUSC5 (Figure 4, Supplementary Figure S11). Also, in mouse limb buds, H3K27ac peaks aligned with DNase I hypersensitivity sites that mark open chromatin (Supplementary Figure S11). These observations suggested that some of the conserved, noncoding elements in the ABR-TUSC5 region could be putative regulatory elements (Supplementary Table S10) that control gene expression in the developing limb.

Identification of putative regulatory elements in the chromosome 17p13.3 region at chr17:900000-1250000 (GRCh37/hg19 assembly). Histone H3K27ac chromatin immunoprecipitation data are shown for human limb buds at embryonic day (E) 33, E41, E44 and E47. Shaded areas highlight peaks of histone H3K27ac modification that align with peaks of evolutionary conservation (based on multiple alignment of the genomes of 100 vertebrates using the PhyloP method) in noncoding regions. The number below each shaded area represents the element number in Supplementary Table 9 that lists the genomic coordinates of the H3K27ac peaks. The histone H3K27ac chromatin immunoprecipitation data were generated by Cotney et al.36 A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
In our population-based study, the prevalence of isolated SHFM without other birth defects was 1.24 per 100 000 live births in NYS. Our study included only isolated cases, whereas other studies of SHFM prevalence also considered cases that had other birth defects (including cases with known syndromes); therefore, our prevalence estimate is in the lower range of 1–9 per 100 000 births previously reported for nonsyndromic and syndromic SHFM combined.2, 3, 4 A study in Manitoba, Canada42 is used here as an example for comparing SHFM prevalence between our study and others. The Manitoba study reported a SHFM prevalence of 5 per 100 000 births between 1957 and 2003.42 This prevalence estimate was higher than ours but the SHFM cases in the Manitoba study included fetal deaths and live births with known syndromes or multiple birth defects, whereas our study was restricted to live births without known syndromes or other birth defects.
Our genome-wide search for CNVs in 25 cases, targeted CNV screening in seven cases and candidate gene sequencing in all 32 cases, resulted in the identification of a novel 17q25 microdeletion in one case (3%), 10q24 microduplications in two cases (6%), 17p13.3 microduplications in three cases (9%) and potentially damaging mutations in the TP63 DNA-binding domain in two cases (6%). Our single case with a de novo 10q24 CNV adds to three other reports of de novo 10q24 microduplications in SHFM.16, 43, 44 In previous reports that had samples sizes ranging from 22 to 72 nonsyndromic SHFM cases or case families, the percentage of cases with 10q24 copy-number gains was 6–23%,43, 45, 46 with 17p13.3 copy-number gains was 18–51%45, 46, 47 and with TP63 mutations was 0–11%.5, 45, 48 These reports did not use population-based case groups, and some reports included case families selected because linkage to 10q2443 or 17p13.345 had been detected previously. Therefore, variation in case ascertainment methods might account for the differences in percentages among previous reports, and between our study and previous reports.
Consistent with previous reports,5 the TP63 mutations detected in SHFM cases in this study were in the DNA-binding domain. Mutations in this domain impair the ability of TP63 protein to bind DNA and regulate transcription.49 The role of TP63 in SHFM may be related to the requirement for TP63 in the stratification of epithelial cells.50 The AER is a band of stratified epithelium at the distal edge of the limb bud,7 and in mouse embryos homozygous for Tp63 with a disrupted DNA-binding domain, the AER appeared poorly stratified and failed to form a distinct epithelial multilayer, and the limbs were truncated or absent at birth.51, 52, 53
The microdeletion at 17q25 affected 11 genes from ARMC7 to GRB2. For two of the genes, SUMO2 and GRB2, there is evidence for a role in limb development. TP63 protein is able to be sumoylated by SUMO2, a protein that effects posttranslational modification, and sumoylation modulates TP63 protein stability.54 Further, a TP63 p.Q634X mutation detected in SHFM5 inhibits TP63 protein sumoylation and negatively affects the ability of TP63 to regulate transcription.55 The binding of GRB2 to FGFR2, a SHFM gene14 that encodes a receptor tyrosine kinase involved in fibroblast growth factor signaling, regulates downstream signaling through FGFR2.56 Possible mechanisms for how the microdeletion leads to SHFM include deleting regulatory regions of SHFM genes, causing haploinsufficiency of SHFM genes, and unmasking damaging, recessive variants in either SHFM genes or SHFM regulatory regions on the other chromosome.
At chromosome 10q24, CNVs overlapped the LBX1-FGF8 region proposed to contain a series of enhancer elements that control FGF8 expression.38, 39, 40 FGF8, a ligand involved in fibroblast growth factor signaling, is expressed in the AER throughout its existence57 and is needed to maintain the AER;58 loss of function of Fgf8 in the AER of mouse embryos results in missing or shortened digits.59 The consequences of rearranging the predicted enhancers relative to their presumed target, Fgf8, have been explored by generating tandem duplications of the Lbx1-Fgf8 region in mouse embryos.40 The duplications placed the Lbx1 promoter at the genomic position usually occupied by Fgf8 resulting in ectopic expression of Lbx1 in structures where Fgf8 was normally expressed, including the AER.40 In the Dactylaplasia mouse model of SHFM, caused by the insertion of retrotransposons in the Lbx1-Fgf8 region, there is ectopic expression of the retrotransposon elements within the AER, cell death of the AER and limb defects similar to SHFM.60, 61 The insertions are approximately 7 kb in length60 and are expected to change the spatial arrangement of regulatory elements in the Lbx1-Fgf8 region relative to target promoters. Thus, findings from the Dactylaplasia mouse model support the concept that modifying the spatial organization of regulatory elements in the LBX1-FGF8 region could be part of the causal mechanism of SHFM due to CNVs at 10q24.40
Copy-number gains at 17p13.3 overlapped the ABR-TUSC5 region. Together, the CNVs, evolutionarily conserved gene order and preliminary evidence for conserved regulatory elements at 17p13.3 prompted us to hypothesize that elements regulating the expression of a limb development gene were located in this region. As proposed for the LBX1-FGF8 region, tampering with the spatial arrangement of the regulatory elements could cause dysregulation of gene expression. The target gene(s) of these putative regulatory elements is unknown, but a candidate is the transcription factor, BHLHA9, based on its location within the ABR-TUSC5 region and on reports linking it to limb development. Homozygous mutations in the BHLHA9 DNA-binding domain cause mesoaxial synostotic syndactyly with phalangeal reduction, Malik-Percin type (Online Mendelian Inheritance in Man 609432), a disorder with a clinical phenotype that includes shortened phalanges, clinodactyly and fusion of toes.62 A homozygous mutation in the DNA-binding domain of BHLHA9 was also detected in a patient whose clinical features of polydactyly, syndactyly, camptodactyly and dysplastic nails, resulted in a diagnosis of complex camptosynpolydactyly (Online Mendelian Inheritance in Man 607539).63 Moreover, in zebrafish embryos, Bhlha9 is expressed in the developing fins, and Bhlha9 knockdown led to shortening of the pectoral fins.45 The finding that Bhlha9-null mice display cutaneous syndactyly because of reduced apoptosis between the digits64 implicates BHLHA9 in at least one aspect of limb development, interdigital apoptosis, but greater definition of the role of BHLHA9 in limb development is needed.
Additional support for the hypothesis that 17p13.3 CNVs lead to SHFM by disturbing the organization of regulatory elements within this region is provided by data showing that 17p13.3 CNVs in SHFM were relatively small in size (mean of 263 kb), overlapped BHLHA9, and had breakpoints in or near the ABR-TUSC5 region.65 By contrast, in individuals who did not have SHFM but were mostly affected with intellectual disability, 17p13.3 duplications were larger (mean size of 1.1 Mb), only sometimes overlapped BHLHA9, and did not interrupt the ABR-TUSC5 region because the breakpoints of these duplications usually fell outside of this region.65 The investigators suggested that BHLHA9 duplication might be necessary for SHFM pathogenesis due to 17p13.3 CNVs but disturbance of regulatory elements near to BHLHA9 was probably also part of the pathogenic mechanism.
Our study had several strengths. The detection of chromosome microduplications and microdeletions in several SHFM cases highlights the importance of including chromosomal microarray testing as part of the diagnostic assessment of SHFM patients. Also, the 10q24 and 17p13.3 CNVs in our cases were similar to those in previous reports of SHFM,16, 17, 43, 44, 45, 66 adding to the evidence that these CNVs cause SHFM. One Iowa case had a de novo 10q24 CNV, confirming previous reports that de novo genetic variants are contributors to some cases of SHFM16, 43, 44, 65 and emphasizing the need to test parental DNA to determine whether potentially causative variants for SHFM are de novo, co-segregate with the phenotype, or show incomplete penetrance. Reports of partially penetrant 10q24 and 17p13.3 CNVs in SHFM67, 68 suggest that other modifier variants or genes also contribute to determining whether SHFM occurs. The finding that manifestation of the SHFM phenotype in the Dactylaplasia mouse model relies not only on insertions in the Lbx1-Fgf8 genomic region but also on being homozygous for a recessive allele at a locus on another chromosome69 further supports the involvement of multiple interacting loci in producing the SHFM phenotype.
This study also had a number of limitations. No medical record data were available to perform a clinical evaluation of cases, and instead, we relied on hospital reporting of birth defects to our registry to assign the British Paediatric Association codes that were used to identify SHFM cases. It is possible that heterogeneity in documenting birth defects among health care institutions and in coding practices among coders affected whether a patient was identified as a SHFM case. In addition, the term ‘ectrodactyly’, used for searching the registry’s narrative case description to identify SHFM, does not describe central ray deficiencies exclusively and may not represent solely SHFM. However, only two of the 25 NYS cases had ‘ectrodactyly’ as the only narrative description of SHFM. Another limitation was that the lack of clinical data on our cases made it difficult to determine whether any had SHFM with long bone deficiency (SHFLD); 17p13.3 microduplications overlapping BHLHA9 have been detected in many previous reports of SHFLD.45, 46, 47 The narrative case description for one of our NYS cases indicated a longitudinal deficiency of the tibula (suggestive of SHFLD) but we did not detect CNVs on chromosome 10 or 17 or mutations in TP63 in this case. Finally, because DNA specimens were not available for the parents of SHFM cases from NYS, we could not determine whether genetic variants arose de novo in those cases.
To conclude, we provided a population-based estimate for the prevalence of isolated SHFM without other major birth defects and detected potentially damaging TP63 mutations and CNVs in 8 of 32 isolated SHFM cases. The 17q25 microdeletion has not been reported previously in SHFM, and two candidate SHFM genes (SUMO2 and GRB2) within the deleted region are worth following up in other populations. The 10q24 and 17p13.3 CNVs were located in genomic regions that share certain characteristics: evolutionarily conserved gene order, putative regulatory elements and a limb development gene locus. Therefore, the concept that CNVs shuffle the arrangement of regulatory elements leading to dysregulation of gene expression and SHFM, previously proposed for the LBX1-FGF8 region,40 might also apply to the ABR-TUSC5 region. Our findings and those of others on microdeletions70 and microduplications40 in SHFM suggest that CNVs can cause SHFM by deleting or disrupting regulatory elements that control gene transcription in the limb bud. Further investigation is needed to understand how dysregulated gene expression during digit development leads to SHFM pathogenesis.
References
Gurrieri, F. & Everman, D. B. Clinical, genetic, and molecular aspects of split-hand/foot malformation: an update. Am. J. Med. Genet. A 161A, 2860–2872 (2013).
Froster-Iskenius, U. G. & Baird, P. A. Limb reduction defects in over one million consecutive livebirths. Teratology 39, 127–135 (1989).
Stoll, C., Calzolari, E., Cornel, M., Garcia-Minaur, S., Garne, E., Nevin, N. et al. A study on limb reduction defects in six European regions. Ann. Genet. 39, 99–104 (1996).
Bedard, T., Lowry, R. B., Sibbald, B., Kiefer, G. N. & Metcalfe, A. Congenital limb deficiencies in Alberta-a review of 33 years (1980-2012) from the Alberta Congenital Anomalies Surveillance System (ACASS). Am. J. Med. Genet. A 167A, 2599–2609 (2015).
van Bokhoven, H., Hamel, B. C., Bamshad, M., Sangiorgi, E., Gurrieri, F., Duijf, P. H. et al. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am. J. Hum. Genet. 69, 481–492 (2001).
Khan, S., Basit, S., Zimri, F. K., Ali, N., Ali, G., Ansar, M. et al. A novel homozygous missense mutation in WNT10B in familial split-hand/foot malformation. Clin. Genet. 82, 48–55 (2012).
Fernandez-Teran, M. & Ros, M. A. The Apical Ectodermal Ridge: morphological aspects and signaling pathways. Int. J. Dev. Biol. 52, 857–871 (2008).
Duijf, P. H., van Bokhoven, H. & Brunner, H. G. Pathogenesis of split-hand/split-foot malformation. Hum. Mol. Genet. 12 (Spec No 1), R51–R60 (2003).
Kelley, R. O. Early development of the vertebrate limb: an introduction to morphogenetic tissue interactions using scanning electron microscopy. Scan. Electron Microsc. Pt 2, 827–836 (1985).
Al-Qattan, M. M. & Kozin, S. H. Update on embryology of the upper limb. J. Hand Surg. Am. 38, 1835–1844 (2013).
Kjaer, K. W., Hansen, L., Schwabe, G. C., Marques-de-Faria, A. P., Eiberg, H., Mundlos, S. et al. Distinct CDH3 mutations cause ectodermal dysplasia, ectrodactyly, macular dystrophy (EEM syndrome). J. Med. Genet. 42, 292–298 (2005).
Shamseldin, H. E., Faden, M. A., Alashram, W. & Alkuraya, F. S. Identification of a novel DLX5 mutation in a family with autosomal recessive split hand and foot malformation. J. Med. Genet. 49, 16–20 (2012).
Simonis, N., Migeotte, I., Lambert, N., Perazzolo, C., de Silva, D. C., Dimitrov, B. et al. FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly. J. Med. Genet. 50, 585–592 (2013).
Barnett, C. P., Nataren, N. J., Klingler-Hoffmann, M., Schwarz, Q., Chong, C. E., Lee, Y. K. et al. Ectrodactyly and lethal pulmonary acinar dysplasia associated with homozygous FGFR2 mutations identified by exome sequencing. Hum. Mutat. 37, 955–963 (2016).
Spielmann, M., Kakar, N., Tayebi, N., Leettola, C., Nurnberg, G., Sowada, N. et al. Exome sequencing and CRISPR/Cas genome editing identify mutations of ZAK as a cause of limb defects in humans and mice. Genome Res. 26, 183–191 (2016).
Kano, H., Kurosawa, K., Horii, E., Ikegawa, S., Yoshikawa, H., Kurahashi, H. et al. Genomic rearrangement at 10q24 in non-syndromic split-hand/split-foot malformation. Hum. Genet. 118, 477–483 (2005).
Armour, C. M., Bulman, D. E., Jarinova, O., Rogers, R. C., Clarkson, K. B., DuPont, B. R. et al. 17p13.3 microduplications are associated with split-hand/foot malformation and long-bone deficiency (SHFLD). Eur. J. Hum. Genet. 19, 1144–1151 (2011).
Theisen, A., Rosenfeld, J. A., Shane, K., McBride, K. L., Atkin, J. F., Gaba, C. et al. Refinement of the region for split hand/foot malformation 5 on 2q31.1. Mol. Syndromol. 1, 262–271 (2010).
Corona-Rivera, A., Corona-Rivera, J. R., Bobadilla-Morales, L., Garcia-Cobian, T. A. & Corona-Rivera, E. Holoprosencephaly, hypertelorism, and ectrodactyly in a boy with an apparently balanced de novo t(2;4) (q14.2;q35). Am. J. Med. Genet. 90, 423–426 (2000).
Correa-Cerro, L., Garciaz-Cruz, D., Diaz-Castanos, L., Figuera, L. E. & Sanchez-Corona, J. Interstitial deletion 6q16.2q22.2 in a child with ectrodactyly. Ann. Genet. 39, 105–109 (1996).
Faiyaz ul Haque, M., Uhlhaas, S., Knapp, M., Schuler, H., Friedl, W., Ahmad, M. et al. Mapping of the gene for X-chromosomal split-hand/split-foot anomaly to Xq26-q26.1. Hum. Genet. 91, 17–19 (1993).
Wang, Y., Sharpe-Stimac, M., Cross, P. K., Druschel, C. M. & Hwang, S. A. Improving case ascertainment of a population-based birth defects registry in New York State using hospital discharge data. Birth Defects Res. A Clin. Mol. Teratol. 73, 663–668 (2005).
Saavedra-Matiz, C. A., Isabelle, J. T., Biski, C. K., Duva, S. J., Sweeney, M. L., Parker, A. L. et al. Cost-effective and scalable DNA extraction method from dried blood spots. Clin. Chem. 59, 1045–1051 (2013).
Yoon, P. W., Rasmussen, S. A., Lynberg, M. C., Moore, C. A., Anderka, M., Carmichael, S. L. et al. The National Birth Defects Prevention Study. Public Health Rep. 116 (Suppl 1), 32–40 (2001).
Wang, K., Li, M., Hadley, D., Liu, R., Glessner, J., Grant, S. F. et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17, 1665–1674 (2007).
Consortium, I. H. The International HapMap Project. Nature 426, 789–796 (2003).
Shaikh, T. H., Gai, X., Perin, J. C., Glessner, J. T., Xie, H., Murphy, K. et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 19, 1682–1690 (2009).
MacDonald, J. R., Ziman, R., Yuen, R. K., Feuk, L. & Scherer, S. W. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 42, D986–D992 (2014).
Harrow, J., Frankish, A., Gonzalez, J. M., Tapanari, E., Diekhans, M., Kokocinski, F. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Hamosh, A., Scott, A. F., Amberger, J. S., Bocchini, C. A. & McKusick, V. A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–D517 (2005).
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P. et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86, 749–764 (2010).
Rosenbloom, K. R., Armstrong, J., Barber, G. P., Casper, J., Clawson, H., Diekhans, M. et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 43, D670–D681 (2015).
Firth, H. V., Richards, S. M., Bevan, A. P., Clayton, S., Corpas, M., Rajan, D. et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am. J. Hum. Genet. 84, 524–533 (2009).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Cotney, J., Leng, J., Yin, J., Reilly, S. K., DeMare, L. E., Emera, D. et al. The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell 154, 185–196 (2013).
DeMare, L. E., Leng, J., Cotney, J., Reilly, S. K., Yin, J., Sarro, R. et al. The genomic landscape of cohesin-associated chromatin interactions. Genome Res. 23, 1224–1234 (2013).
Beermann, F., Kaloulis, K., Hofmann, D., Murisier, F., Bucher, P. & Trumpp, A. Identification of evolutionarily conserved regulatory elements in the mouse Fgf8 locus. Genesis 44, 1–6 (2006).
Komisarczuk, A. Z., Kawakami, K. & Becker, T. S. Cis-regulation and chromosomal rearrangement of the fgf8 locus after the teleost/tetrapod split. Dev. Biol. 336, 301–312 (2009).
Marinic, M., Aktas, T., Ruf, S. & Spitz, F. An integrated holo-enhancer unit defines tissue and gene specificity of the Fgf8 regulatory landscape. Dev. Cell 24, 530–542 (2013).
Creyghton, M. P., Cheng, A. W., Welstead, G. G., Kooistra, T., Carey, B. W., Steine, E. J. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21931–21936 (2010).
Elliott, A. M., Reed, M. H., Chudley, A. E., Chodirker, B. N. & Evans, J. A. Clinical and epidemiological findings in patients with central ray deficiency: split hand foot malformation (SHFM) in Manitoba, Canada. Am. J. Med. Genet. A 140, 1428–1439 (2006).
Everman, D. B., Morgan, C. T., Lyle, R., Laughridge, M. E., Bamshad, M. J., Clarkson, K. B. et al. Frequency of genomic rearrangements involving the SHFM3 locus at chromosome 10q24 in syndromic and non-syndromic split-hand/foot malformation. Am. J. Med. Genet. A 140, 1375–1383 (2006).
Dimitrov, B. I., de Ravel, T., Van Driessche, J., de Die-Smulders, C., Toutain, A., Vermeesch, J. R. et al. Distal limb deficiencies, micrognathia syndrome, and syndromic forms of split hand foot malformation (SHFM) are caused by chromosome 10q genomic rearrangements. J. Med. Genet. 47, 103–111 (2010).
Klopocki, E., Lohan, S., Doelken, S. C., Stricker, S., Ockeloen, C. W., Soares Thiele de Aguiar, R. et al. Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion. J. Med. Genet. 49, 119–125 (2012).
Nagata, E., Kano, H., Kato, F., Yamaguchi, R., Nakashima, S., Takayama, S. et al. Japanese founder duplications/triplications involving BHLHA9 are associated with split-hand/foot malformation with or without long bone deficiency and Gollop-Wolfgang complex. Orphanet J. Rare Dis. 9, 125 (2014).
Petit, F., Jourdain, A. S., Andrieux, J., Baujat, G., Baumann, C., Beneteau, C. et al. Split hand/foot malformation with long-bone deficiency and BHLHA9 duplication: report of 13 new families. Clin. Genet. 85, 464–469 (2014).
de Mollerat, X. J., Everman, D. B., Morgan, C. T., Clarkson, K. B., Rogers, R. C., Colby, R. S. et al. P63 mutations are not a major cause of non-syndromic split hand/foot malformation. J. Med. Genet. 40, 55–61 (2003).
Celli, J., Duijf, P., Hamel, B. C., Bamshad, M., Kramer, B., Smits, A. P. et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 99, 143–153 (1999).
Koster, M. I., Kim, S., Mills, A. A., DeMayo, F. J. & Roop, D. R. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 18, 126–131 (2004).
Lo Iacono, N., Mantero, S., Chiarelli, A., Garcia, E., Mills, A. A., Morasso, M. I. et al. Regulation of Dlx5 and Dlx6 gene expression by p63 is involved in EEC and SHFM congenital limb defects. Development 135, 1377–1388 (2008).
Mills, A. A., Zheng, B., Wang, X. J., Vogel, H., Roop, D. R. & Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398, 708–713 (1999).
Yang, A., Schweitzer, R., Sun, D., Kaghad, M., Walker, N., Bronson, R. T. et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398, 714–718 (1999).
Ghioni, P., D'Alessandra, Y., Mansueto, G., Jaffray, E., Hay, R. T., La Mantia, G. et al. The protein stability and transcriptional activity of p63alpha are regulated by SUMO-1 conjugation. Cell Cycle 4, 183–190 (2005).
Huang, Y. P., Wu, G., Guo, Z., Osada, M., Fomenkov, T., Park, H. L. et al. Altered sumoylation of p63alpha contributes to the split-hand/foot malformation phenotype. Cell Cycle 3, 1587–1596 (2004).
Lin, C. C., Melo, F. A., Ghosh, R., Suen, K. M., Stagg, L. J., Kirkpatrick, J. et al. Inhibition of basal FGF receptor signaling by dimeric Grb2. Cell 149, 1514–1524 (2012).
Crossley, P. H. & Martin, G. R. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development 121, 439–451 (1995).
Moon, A. M. & Capecchi, M. R. Fgf8 is required for outgrowth and patterning of the limbs. Nat. Genet. 26, 455–459 (2000).
Lewandoski, M., Sun, X. & Martin, G. R. Fgf8 signalling from the AER is essential for normal limb development. Nat. Genet. 26, 460–463 (2000).
Kano, H., Kurahashi, H. & Toda, T. Genetically regulated epigenetic transcriptional activation of retrotransposon insertion confers mouse dactylaplasia phenotype. Proc. Natl Acad. Sci. USA 104, 19034–19039 (2007).
Seto, M. L., Nunes, M. E., MacArthur, C. A. & Cunningham, M. L. Pathogenesis of ectrodactyly in the Dactylaplasia mouse: aberrant cell death of the apical ectodermal ridge. Teratology 56, 262–270 (1997).
Malik, S., Percin, F. E., Bornholdt, D., Albrecht, B., Percesepe, A., Koch, M. C. et al. Mutations affecting the BHLHA9 DNA-binding domain cause MSSD, mesoaxial synostotic syndactyly with phalangeal reduction, Malik-Percin type. Am. J. Hum. Genet. 95, 649–659 (2014).
Phadke, S. R., Kar, A., Bhowmik, A. D. & Dalal, A. Complex camptosynpolydactyly and mesoaxial synostotic syndactyly with phalangeal reduction are allelic disorders. Am. J. Med. Genet. A 170, 1622–1625 (2016).
Schatz, O., Langer, E. & Ben-Arie, N. Gene dosage of the transcription factor Fingerin (bHLHA9) affects digit development and links syndactyly to ectrodactyly. Hum. Mol. Genet. 23, 5394–5401 (2014).
Curry, C. J., Rosenfeld, J. A., Grant, E., Gripp, K. W., Anderson, C., Aylsworth, A. S. et al. The duplication 17p13.3 phenotype: analysis of 21 families delineates developmental, behavioral and brain abnormalities, and rare variant phenotypes. Am. J. Med. Genet. A 161A, 1833–1852 (2013).
Al Kaissi, A., Ganger, R., Rotzer, K. M., Klaushofer, K. & Grill, F. A child with split-hand/foot associated with tibial hemimelia (SHFLD syndrome) and thrombocytopenia maps to chromosome region 17p13.3. Am. J. Med. Genet. A 164A, 2338–2343 (2014).
Filho, A. B., Souza, J., Faucz, F. R., Sotomaior, V. S., Dupont, B., Bartel, F. et al. Somatic/gonadal mosaicism in a syndromic form of ectrodactyly, including eye abnormalities, documented through array-based comparative genomic hybridization. Am. J. Med. Genet. A 155A, 1152–1156 (2011).
Petit, F., Andrieux, J., Demeer, B., Collet, L. M., Copin, H., Boudry-Labis, E. et al. Split-hand/foot malformation with long-bone deficiency and BHLHA9 duplication: two cases and expansion of the phenotype to radial agenesis. Eur. J. Med. Genet. 56, 88–92 (2013).
Johnson, K. R., Lane, P. W., Ward-Bailey, P. & Davisson, M. T. Mapping the mouse dactylaplasia mutation, Dac, and a gene that controls its expression, mdac. Genomics 29, 457–464 (1995).
Rattanasopha, S., Tongkobpetch, S., Srichomthong, C., Kitidumrongsook, P., Suphapeetiporn, K. & Shotelersuk, V. Absent expression of the osteoblast-specific maternally imprinted genes, DLX5 and DLX6, causes split hand/split foot malformation type I. J. Med. Genet. 51, 817–823 (2014).
Acknowledgements
We thank Matthew Shudt and Zhen Zhang at the Wadsworth Center Applied Genomics Technologies Core, New York State Department of Health, for next-generation sequencing; April Atkins and Emily McGrath at the Wadsworth Center, New York State Department of Health, for technical assistance; and Nathan Pankratz, University of Minnesota, and Karl G. Hill, Social Development Research Group, University of Washington, for generously sharing population B-allele frequency and GC content files for PennCNV software. This work was funded by the Intramural Research Program of the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development (Contracts HHSN275201100001I, HHSN27500005 and N01-DK-73431) and the Iowa Center for Birth Defects Research and Prevention (5U01DD001035).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Carter, T., Sicko, R., Kay, D. et al. Copy-number variants and candidate gene mutations in isolated split hand/foot malformation. J Hum Genet 62, 877–884 (2017). https://doi.org/10.1038/jhg.2017.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.56
This article is cited by
-
A new case of 17p13.3p13.1 microduplication resulted from unbalanced translocation: clinical and molecular cytogenetic characterization
Molecular Cytogenetics (2021)
-
Little to Give, Much to Gain—What Can You Do With a Dried Blood Spot?
Current Environmental Health Reports (2020)
-
Split hand/foot malformation with long bone deficiency associated with BHLHA9 gene duplication: a case report and review of literature
BMC Medical Genetics (2019)
-
Comprehensive clinical and molecular studies in split-hand/foot malformation: identification of two plausible candidate genes (LRP6 and UBA2)
European Journal of Human Genetics (2019)
-
Duplication of 10q24 locus: broadening the clinical and radiological spectrum
European Journal of Human Genetics (2019)
