
Abstract
Marfan syndrome is an autosomal dominant disorder affecting mainly the skeletal, ocular and cardiovascular systems. Most cases are caused by mutations in the fibrillin-1 gene (FBN1), although there are some reports on deletions involving FBN1 and other additional genes. We report a male patient who was first evaluated at 4 years of age. Echocardiogram showed a mildly dilated aortic sinus. He also had a history of muscular ventral septal defect which was closed spontaneously and trivial mitral regurgitation. Other phenotypic features include frontal bossing, anteverted ears, joint hyperlaxity, learning disability, skin striae, and height and weight in the >97th centile but no other diagnostic findings of MFS and does not fulfill the revised Ghent criteria. Chromosomal microarray analysis showed a deletion of approximately 36.8 kb at 15q21.1, which starts in intron 6 and ends in intron 9 and includes three FBN1 exons. Sequence analysis of the breakpoint region confirmed the deletion and revealed a concomitant insertion of a retrotransposon within the intron 6/intron 9 region. The intragenic deletion of exons 7–9 was likely the result of a retrotransposition event by a MAST2-SVA element mediated by repetitive sequences.
Similar content being viewed by others

Introduction
Marfan syndrome (MFS; MIM #154700), an autosomal dominant connective tissue disorder, is mainly caused by mutations in the fibrillin-1 gene (FBN1) and affects the skeletal, ocular and cardiovascular systems.1 There is locus heterogeneity in that mutations in the genes encoding transforming growth factor beta-receptors I and II (TGFBR1 and TGFBR2) have been shown to cause MFS-related disorders like MFS type II,2 Loeys–Dietz syndrome3 and thoracic aortic aneurysms and dissections.4 To date, over 3000 FBN1 mutations have been reported in the FBN1 Universal Mutation Database (UMD, http://www.umd.be/FBN1/) with point mutations accounting for 73%, and large rearrangements for <2% of the reported mutations. Of the point mutations, 80% were missense mutations. The type and location of FBN1 mutations are known to correlate with some phenotypic features. For example, neonatal MFS is usually linked to FBN1 mutations located between exons 23–325 and missense mutations involving cysteine have been associated with ectopia lentis.6
It has been estimated that about 35% of the genome is derived from retrotransposed sequences,7 with about 0.27% of human genetic diseases caused by retrotransposable elements (RE).8 The three major types of RE are LINE-1 (long interspersed nucleotide element-1) or L1, Alu and SVA (SINE-VNTR-Alu) and they comprise 17, 11 and 0.2% of the human genome mass, respectively.9 REs have been shown to influence genome stability and retrotransposition has been shown to cause genetic diseases and cancer.10 L1 is the most abundant LINE element but most copies are inactive and only 80–100 elements are currently active in any individual.11 SVAs are composite transposons comprising SINE-R, VNTR and Alu elements. L1 is the only autonomous retrotransposon and has also been involved in retrotransposition of the other non-autonomous elements like Alu and SVA.12, 13 In this study, we report for the first time a retrotransposable element (RE) within the FBN1 gene concomitant with the deletion of three exons in a child with mildly dilated aortic sinus.
Materials and methods
The child first presented at 4 years of age in the Genetics Clinic, with a history of muscular ventricular septal defect and trivial mitral regurgitation, and echocardiogram findings of a mildly dilated aortic sinus. No family history is available. He also had learning disability and attention deficit hyperactivity disorder. His height was 106 cm (75th–90th centile), his weight was 15.9 kg (50th–75th centile) and his head circumference was 52.5 cm (97th centile). Initial clinical examination showed frontal bossing, downslanting palpebral fissures, anteverted ears, joint hypermobility and bilateral 5th finger clinodactyly. He did not have palate or chest wall abnormalities or flat feet. Karyotype and Fragile X testing returned normal results.
From 6 years of age to the present, his height, weight and head circumference increased to above the 97th centile. He has no cardiac symptoms and his latest echocardiogram showed a prominent aortic sinus (Z-score +0.73). Psychological assessment done at 7 years of age showed an IQ of 60 and he has been attending a special school since. He developed obstructive sleep apnea (OSA), most probably related to his excessive weight gain, and required tonsillectomy and adenoidectomy at 11 years of age. At his most recent clinic visit when he was 15 years old, his height was 182.4 cm (>97th centile), his weight was 98 kg (>97th centile) and his body mass index (BMI) was 29.5. Apart from skin striae, he had no other systemic features of Marfan syndrome. Based on the revised Ghent criteria,14 his systemic score was 1. Eye examination showed no ectopia lentis. Therefore, he does not fulfill the revised Ghent criteria for diagnosis of Marfan syndrome.
Study approval for the investigation of submicroscopic chromosomal abnormalities was granted by the SingHealth Institutional Review Board which oversees all research activities in the hospital. Peripheral blood sample was collected with written informed consent. Chromosomal microarray analysis (CMA) using the Agilent 1 M array kit (Agilent Technologies Inc., Santa Clara, CA, USA) was performed according to manufacturer’s guidelines. DNA from the patient was hybridized against the male human genomic DNA reference from Promega (Promega Corp., Madison, WI, USA). Post-hybridization, the slide was scanned with Agilent G2505C scanner at 5 μm resolution. Results were analyzed using the Agilent Genomic Workbench Lite software (version 6.5) with the ADM-2 algorithm.
Breakpoint analysis was performed by long-range PCR followed by Sanger sequencing. Long-range PCR was carried out using the TaKaRa PrimeStar GXL kit (TaKaRa Bio Inc., Shiga, Japan). PCR products were sequenced using the ABI BigDye Terminator (v3.1) and the ABI 3130 Genetic Analyser (Applied Biosystems, Foster City, CA, USA). Sequences obtained were analyzed using publicly available NCBI Blast, UCSC BLAT the sequence alignment tool on UCSC (http://genome.ucsc.edu/cgi-bin/hgBlat?) and RepeatMasker (http://www.repeatmasker.org) for interspersed repeat sequences.
Results
Chromosome microarray analysis revealed a deletion of at least 36.8 kb at 15q21.1 (Figure 1) from chr15:48 815 355–48 852 135 (hg19). Long-range PCR followed by DNA sequencing showed that the exact breakpoints were at chr15:48 814 550 and chr15:48 853 211 (Figure 2a and b). The deletion spans 38 662 bp with exons 7−9 of the FBN1 gene completely deleted. The telomeric breakpoint occurred in intron 6 and the centromeric breakpoint was in intron 9 of the gene. It is not known if the deletion was inherited or had occurred de novo as DNA samples from biological parents were not available for analysis.

CMA result showing the deletion at 15q21.1 as depicted in Genomic Workbench Lite software. A full color version of this figure is available at the Journal of Human Genetics journal online.

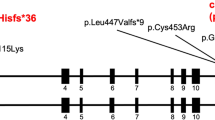
Depiction of the deletion and the sequences at the breakpoints. (a) The vertical arrows in the top figure show the approximate position of the breakpoints in introns 6 and 9 of the FBN1 gene and the bottom figure shows the structure of the MAST2-SVA element. The two arrows indicate the approximate positions of the primers used to amplify the insert for dideoxy sequencing. The MAST2-SVA is divided into MAST2 (violet), Alu-like (red), VNTR (green), SINE-R (blue), polyadenylation signal (black) and poly(A) tail (orange) based on sequence information in c. (b) Chromatograms and nucleotide sequence at the telomeric (top) and centromeric (bottom) breakpoints showing the comparison between the wild-type FBN1 sequence and the mutant sequence in the patient. (c) Sequence of the MAST2-SVA element, which is inserted at the intron 6 and intron 9 breakpoints of the FBN1 gene in the patient. Nucleotides are shown in the respective colors of the various sections of the SVA (refer to the various regions shown in the bottom figure of a). A full color version of this figure is available at the Journal of Human Genetics journal online.
Long-range PCR using primers from intron 6 and intron 9 (Figure 2a) resulted in a fragment of ~2.5 kb. On the basis of the positions of the primers and the FBN1 sequences at the breakpoints, the inserted element is estimated to be 1.5 kb. Analysis of the sequence at the breakpoint junction using RepeatMasker showed that the inserted sequence had high similarity with the SVA_F retrotransposable elements. Due to a long poly (A) tract, sequencing results were only obtained for ~950 bp from the centromeric breakpoint. A detailed analysis showed that it had the structure of a newly described subfamily called MAST2-SVA. The inserted transposon in the breakpoint junction comprised sequence from exon 1 of the MAST2 gene, an Alu-like fragment, VNTR section, SINE-R sequence and a polyA tail (Figure 2c). Alignment of the sequence using BLAT showed 99.0% similarity to a sequence (Supplementary Figure S1) from intron 1 of the COL23A1 gene on chromosome 5 (chr5: 177944877–177945942, hg19). This sequence was identified as a SVA_F element by Repeat Masker and also included the MAST2 exon 1 sequence. RepeatMasker analysis of the sequence around the breakpoints revealed the presence of a MER1A, family hAT-Charlie element at the intron 9 breakpoint and an MIRb element at the intron 6 breakpoint.
Discussion
The vast majority of MFS cases are caused by point mutations in the FBN1 gene with only an estimated 1–2% of cases due to large intragenic deletions or chromosomal imbalances. Interstitial deletions involving 15q21 and the FBN1 gene are very rare. There have been at least 18 reports of MFS patients with deletions of the whole FBN1 gene and with variable phenotypic presentations varying from mild features of MFS15, 16, 17, 18 to the classical MFS phenotype.19, 20 Two of these patients with gross chromosomal deletions have intellectual disability as well and this has been attributed to deletions of other genes in the region.15, 16 Interestingly, our patient has learning disability and attention deficit hyperactivity disorder despite not having any other neighboring gene deleted. Another patient with a cytogenetic abnormality involving this region had azoospermia and Marfan syndrome. He had an abnormal karyotype with an interstitial chromosome 15 deletion and a supernumerary marker chromosome with the ‘deleted’ chromosome 15 material.21
Fibrillin is a large glycoprotein that is ubiquitous in connective tissues and is the major component of microfibrils. Fibrillin contains 47 epidermal growth factor-like (EGF-like) domains and seven transforming growth factor-like (TGF-β like) domains. Forty-three of the EGF-like domains have a calcium-binding consensus sequence.22 Exons 7 and 8 of FBN1 code for the first two calcium-binding EGF (cbEGF-like) motifs and exon 9 and part of exon 10 code for a TGF-β-like motif.23 The deletion of exons 7 and 8 is predicted to lead to the loss of the first two cbEGF-like motifs and the deletion of exon 9 is predicted to cause a disruption of the TGF-β-like motif. The cbEGF domains are highly conserved and are important for correct folding of the fibrillin protein.24 The inserted SVA contains several acceptor and donor splice sites which may further disrupt the coding sequence of the mRNA.
Large intragenic deletions of FBN1 involving single and multiple exons represent <2% of reported cases of MFS patients, and some of these patients have classical or severe MFS phenotypes.25, 26 None of the previous cases involved deletion of exons 7–9 as found in our patient. The largest reported case was for 37 exons from exons 13–49. Despite it being mosaic, the patient had severe MFS that was clinically diagnosed when she was 3 years old.27 Singh et al.26 described another patient with severe juvenile onset cardiovascular phenotype who had a 10.5 kb genomic deletion from exons 58–63, which the authors postulated would have disrupted cbEGF-like domain functions. Other cases with deletions of exons that are more 3′ also had severe MFS.27, 28 In contrast, our patient with deletion of exons 7−9 does not fulfill the revised Ghent criteria for diagnosis of MFS.
Retrotransposition has been shown to cause insertions, deletions and rearrangements and can also affect gene expression by affecting promoters, splice sites, polyadenylation signals and silencing.9 Retrotranspositional insertion of L1, Alu, SVA elements have been shown to result in single gene disorders and over a hundred such instances have been reported.29 The spectrum of diseases caused by retrotransposition events include congenital muscular dystrophy,30 leukemia,31 CHARGE syndrome32 and neurofibromatosis type 1.33, 34 Notably, none has been reported for FBN1 and Marfan-related syndromes.
SVAs are composite transposons that are mobilized by L1 elements. Its size in our patient is similar to the two cases reported by Vogt et al.33 at for NF1 (1.3 and 1.7 kb). Analysis of the breakpoint junction sequence in our patient showed the insertion of a MAST2-SVA element with a minimum size of 951 bp between intron 6 and intron 9 of the FBN1 gene. As far as we are aware, this is the first instance of a retrotransposon inserting into the FBN1 gene resulting in the loss of several exons. The MAST2-SVA subfamily likely originated from one single source element by an alternative splicing event where the first exon of the MAST2 gene spliced into an SVA, which subsequently retrotransposed.35 This subfamily has been referred to as SVA_F1,35 CpG-SVA36 or MAST2-SVA.37 At least 84 members of this subfamily have been identified, with sizes ranging from 662 to 4255 bp and variable lengths of the component parts, and it is postulated that the CpG-rich portion of exon 1 of the MAST2 gene may play a role as a transcriptional regulator.38 The MAST2-SVA retrotransposon has also been reported in three unrelated patients with leukemia caused by the deletion of the entire HLA-A gene accompanied by the insertion of the retrotransposons.31, 37 The sequence similarity between the inserted sequence in our patient and a MAST2-SVA retrotransposons in intron 1 of COL23A1 gene suggests a possible source of the retrotransposon in our patient. Examination of the breakpoint regions showed the presence of a MIRb element at the intron 6 breakpoint and a MER1A, family hAT-Charlie element at the intron 9 breakpoint. These repetitive elements could have predisposed this region to the retrotransposition event by the MAST2-SVA element.
In conclusion, we describe a patient with mildly dilated aortic sinus but no other diagnostic findings of MFS, and a deletion of exons 7–9 of the FBN1 gene that was the result of a retrotransposition event by a MAST2-SVA element. The patient does not meet the Ghent criteria for MFS but may have haploinsufficiency and disruption of the FBN1 gene.
References
De Paepe, A., Devereux, R. B., Dietz, H. C., Hennekam, R. C. & Pyeritz, R. E . Revised diagnostic criteria for the Marfan syndrome. Am. J. Med. Genet. 62, 417–426 (1996).
Mizuguchi, T., Collod-Beroud, G., Akiyama, T., Abifadel, M., Harada, N., Morisaki, T. et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet. 36, 855–860. (2004).
Loeys, B. L., Chen, J., Neptune, E. R., Judge, D. P., Podowski, M., Holm, T. et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 37, 275–281. (2005).
Pannu, H., Tran-Fadulu, V. & Milewicz, D. M . Genetic basis of thoracic aortic aneurysms and aortic dissections. Am. J. Med. Genet. C Semin. Med. Genet. 139C, 10–16. (2005).
Putnam, E. A., Cho, M., Zinn, A. B., Towbin, J. A., Byers, P. H. & Milewicz, D. M . Delineation of the Marfan phenotype associated with mutations in exons 23-32 of the FBN1 gene. Am. J. Med. Genet. 62, 233–242 (1996).
Faivre, L., Collod-Beroud, G., Loeys, B. L., Child, A., Binquet, C., Gautier, E. et al. Effect of mutation type and location on clinical outcome in 1013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am. J. Hum. Genet. 81, 454–466. (2007).
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921. (2001).
Callinan, P. A. & Batzer, M. A . Retrotransposable elements and human disease. Genome Dyn. 1, 104–115. (2006).
Kaer, K. & Speek, M . Retroelements in human disease. Gene 518, 231–241. (2013).
Solyom, S. & Kazazian, H. H. Jr. Mobile elements in the human genome: implications for disease. Genome Med. 4, 12. (2012).
Brouha, B., Schustak, J., Badge, R. M., Lutz-Prigge, S., Farley, A. H., Moran, J. V. et al. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl Acad. Sci. USA 100, 5280–5285. (2003).
Hancks, D. C., Goodier, J. L., Mandal, P. K., Cheung, L. E. & Kazazian, H. H. Jr. Retrotransposition of marked SVA elements by human L1s in cultured cells. Hum. Mol. Genet. 20, 3386–3400. (2011).
Raiz, J., Damert, A., Chira, S., Held, U., Klawitter, S., Hamdorf, M. et al. The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 40, 1666–1683. (2012).
Loeys, B. L., Dietz, H. C., Braverman, A. C., Callewaert, B. L., De Backer, J., Devereux, R. B. et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 47, 476–485. (2010).
Ades, L. C., Sullivan, K., Biggin, A., Haan, E. A., Brett, M., Holman, K. J. et al. FBN1, TGFBR1, and the Marfan-craniosynostosis/mental retardation disorders revisited. Am. J. Med. Genet. A 140, 1047–1058. (2006).
Hutchinson, S., Furger, A., Halliday, D., Judge, D. P., Jefferson, A., Dietz, H. C. et al. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum. Mol. Genet. 12, 2269–2276. (2003).
Faivre, L., Khau Van Kien, P., Callier, P., Ruiz-Pallares, N., Baudoin, C., Plancke, A. et al. De novo 15q21.1q21.2 deletion identified through FBN1 MLPA and refined by 244 K array-CGH in a female teenager with incomplete Marfan syndrome. Eur J. Med. Genet. 53, 208–212. (2010).
Dordoni, C., Ciaccio, C., Santoro, G., Venturini, M., Cavallari, U., Ritelli, M. et al. Marfan syndrome: report of a complex phenotype due to a 15q21.1 contiguos gene deletion encompassing FBN1, and literature review. Am. J. Med. Genet. A 173, 200–206. (2016).
Hilhorst-Hofstee, Y., Hamel, B. C., Verheij, J. B., Rijlaarsdam, M. E., Mancini, G. M., Cobben, J. M. et al. The clinical spectrum of complete FBN1 allele deletions. Eur. J. Hum. Genet. 19, 247–252. (2011).
Colovati, M. E., da Silva, L. R., Takeno, S. S., Mancini, T. I., N Dutra, A. R., Guilherme, R. S. et al. Marfan syndrome with a complex chromosomal rearrangement including deletion of the FBN1 gene. Mol. Cytogenet. 5, 5. (2012).
Quinonez, S. C., Gelehrter, T. D. & Uhlmann, W. R . A Marfan syndrome-like phenotype caused by a neocentromeric supernumerary ring chromosome 15. Am. J. Med. Genet. A 173, 268–273. (2016).
Corson, G. M., Chalberg, S. C., Dietz, H. C., Charbonneau, N. L. & Sakai, L. Y . Fibrillin binds calcium and is coded by cDNAs that reveal a multidomain structure and alternatively spliced exons at the 5′ end. Genomics 17, 476–484. (1993).
Saharinen, J. & Keski-Oja, J . Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Biol. Cell 11, 2691–2704. (2000).
Aoyama, T., Tynan, K., Dietz, H. C., Francke, U. & Furthmayr, H . Missense mutations impair intracellular processing of fibrillin and microfibril assembly in Marfan syndrome. Hum. Mol. Genet. 2, 2135–2140. (1993).
Furtado, L. V., Wooderchak-Donahue, W., Rope, A. F., Yetman, A. T., Lewis, T., Plant, P. et al. Characterization of large genomic deletions in the FBN1 gene using multiplex ligation-dependent probe amplification. BMC Med. Genet. 12, 119. (2011).
Singh, K. K., Elligsen, D., Liersch, R., Schubert, S., Pabst, B., Arslan-Kirchner, M. et al. Multi-exon out of frame deletion of the FBN1 gene leading to a severe juvenile onset cardiovascular phenotype in Marfan syndrome. J. Mol. Cell Cardiol. 42, 352–356. (2007).
Blyth, M., Foulds, N., Turner, C. & Bunyan, D . Severe Marfan syndrome due to FBN1 exon deletions. Am. J. Med. Genet. A 146A, 1320–1324. (2008).
Liu, W., Schrijver, I., Brenn, T., Furthmayr, H. & Francke, U . Multi-exon deletions of the FBN1 gene in Marfan syndrome. BMC Med. Genet. 2, 11. (2001).
Hancks, D. C. & Kazazian, H. H. Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 7, 9. (2016).
Xiong, H., Wang, S., Kobayashi, K., Jiang, Y., Wang, J., Chang, X. et al. Fukutin gene retrotransposal insertion in a non-Japanese Fukuyama congenital muscular dystrophy (FCMD) patient. Am. J. Med. Genet. A 149A, 2403–2408. (2009).
Takasu, M., Hayashi, R., Maruya, E., Ota, M., Imura, K., Kougo, K. et al. Deletion of entire HLA-A gene accompanied by an insertion of a retrotransposon. Tissue Antigens 70, 144–150. (2007).
Udaka, T., Okamoto, N., Aramaki, M., Torii, C., Kosaki, R., Hosokai, N. et al. An Alu retrotransposition-mediated deletion of CHD7 in a patient with CHARGE syndrome. Am. J. Med. Genet. A 143A, 721–726. (2007).
Vogt, J., Bengesser, K., Claes, K. B., Wimmer, K., Mautner, V. F., van Minkelen, R. et al. SVA retrotransposon insertion-associated deletion represents a novel mutational mechanism underlying large genomic copy number changes with non-recurrent breakpoints. Genome Biol. 15, R80. (2014).
Wimmer, K., Callens, T., Wernstedt, A. & Messiaen, L . The NF1 gene contains hotspots for L1 endonuclease-dependent de novo insertion. PLoS Genet. 7, e1002371. (2011).
Damert, A., Raiz, J., Horn, A. V., Lower, J., Wang, H., Xing, J. et al. 5′-Transducing SVA retrotransposon groups spread efficiently throughout the human genome. Genome Res. 19, 1992–2008. (2009).
Bantysh, O. B. & Buzdin, A. A . Novel family of human transposable elements formed due to fusion of the first exon of gene MAST2 with retrotransposon SVA. Biochemistry (Mosc) 74, 1393–1399. (2009).
Hancks, D. C., Ewing, A. D., Chen, J. E., Tokunaga, K. & Kazazian, H. H. Jr. Exon-trapping mediated by the human retrotransposon SVA. Genome Res. 19, 1983–1991. (2009).
Zabolotneva, A. A., Bantysh, O., Suntsova, M. V., Efimova, N., Malakhova, G. V., Schumann, G. G. et al. Transcriptional regulation of human-specific SVAF(1) retrotransposons by cis-regulatory MAST2 sequences. Gene 505, 128–136. (2012).
Acknowledgements
This work was funded by grants BMRC 06/1/50/19/485 from the Biomedical Research Council, Agency for Science, Technology and Research, Republic of Singapore and NMRC/CG/006/2013 from the National Medical Research Council, Ministry of Health, Republic of Singapore. We thank Dustin Hancks and Haig Kazazian for helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Brett, M., Korovesis, G., Lai, A. et al. Intragenic multi-exon deletion in the FBN1 gene in a child with mildly dilated aortic sinus: a retrotransposal event. J Hum Genet 62, 711–715 (2017). https://doi.org/10.1038/jhg.2017.32
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.32
This article is cited by
-
Retrotransposon insertion as a novel mutational cause of spinal muscular atrophy
Human Genetics (2023)
-
LINE-1 ORF1p does not determine substrate preference for human/orangutan SVA and gibbon LAVA
Mobile DNA (2020)
-
High MAST2 mRNA expression and its role in diagnosis and prognosis of liver cancer
Scientific Reports (2019)

{kind=link}