
Abstract
Four causative genes, including solute carrier family 20 member 2 (SLC20A2), platelet-derived growth factor receptor b (PDGFRB), platelet-derived growth factor b (PDGFB)and xenotropic and polytropic retrovirus receptor 1 (XPR1), have been identified to cause primary familial brain calcification (PFBC). However, PDGFRB mutations seem to be quite rare and no PDGFRB mutations have been reported in Chinese PFBC patients. A total of 146 PFBC patients including 12 families and 134 sporadic patients were recruited in this study. All of them were previously tested negative for the SLC20A2. Mutational analyses of the entire exons and exon–intron boundaries of PDGFRB were carried out by direct gene sequencing. In silico analyses of the identified variants were conducted using Mutation Taster, PolyPhen-2 and Sorts Intolerant From Tolerant. Two heterozygous variants, c.3G>A and c.2209G>A, of the PDGFRB gene were revealed in two PFBC families, respectively. These two variants were not observed in 200 healthy controls. The variant c.3G>A was located in exon 2 and affected the initiation codon of the PDGFRB gene. The variant c.2209G>A resulted in amino-acid substitutions of aspartic acid to asparagine at position 737. Both of these two variants co-segregated with the disease phenotype (variant carriers in Family 1: I1, II2 and II3; variant carriers in Family 2: I2 and II8), suggesting a pathogenic impact of these variants. The prevalence of PDGFRB mutations in Chinese PFBC patients seems to be quite low, indicating that PDGFRB is not a major causative gene of PFBC in Chinese population.
Similar content being viewed by others


Introduction
Primary familial brain calcification (PFBC), previously known as idiopathic basal ganglia calcification, or Fahr’s disease, is a rare neurological disorder characterized by bilateral accumulation of calcium in the basal ganglia, cerebellum, thalamus and other brain regions.1 Patients with PFBC present a wide spectrum of clinical features, ranging from asymptomatic to diverse disorders, including parkinsonism, dystonia, chorea and psychiatric symptoms.2 For this reason, the diagnosis of PFBC is mainly based on neuroimaging such as cerebral computed tomography (CT) scans. Typically, PFBC is inherited in an autosomal-dominant pattern. To date, four causative genes, including solute carrier family 20 member 2 (SLC20A2), platelet-derived growth factor receptor b (PDGFRB), platelet-derived growth factor b (PDGFB) and xenotropic and polytropic retrovirus receptor 1 (XPR1) have been identified to be responsible for PFBC.3, 4, 5, 6 Among these genes, SLC20A2 encoding type III sodium-dependent phosphate transporter 2 (PiT-2), is the first reported gene and is also known as the most common causative gene of PFBC. Mutations in SLC20A2 have been identified in >40 families worldwide.7 Conversely, the overall mutation spectrum of PDGFRB, PDGFB and XPR1 remains fragmentary. Especially, PDGFRB mutations seem to be quite rare. Until recently, only four mutations of PDGFRB have been reported in one family and three sporadic patients.3, 8, 9 To our best knowledge, no PDGFRB mutations have been reported in Chinese patients with PFBC. Herein, we aim to identify PDGFRB mutations in a large cohort of Chinese PFBC patients and expand the mutation spectrum of PDGFRB.
Materials and methods
Ethical standards
The study complied with the Declaration of Helsinki Principles and was approved by the ethics committee of the First Affiliated Hospital of Fujian Medical University. Written informed consents were obtained from each subject in this study.
Subjects
A total of 146 patients including 12 families and 134 sporadic patients were recruited from multiple hospitals in China from January 2013 to December 2015 in this study. Clinical diagnosis of PFBC was made with the following criteria: (A) Bilateral calcification of the basal ganglia and/or cerebellum, the brain stem, the centrum semiovale, thalamus and the subcortical white matter visualized on neuroimaging (brain CT scan); (B) Absence of biochemical abnormalities, including serum concentration of calcium, phosphate, calcitonin and parathyroid hormone; and (C) Absence of an infectious, toxic or traumatic cause. We excluded the individuals with mild calcification in the same ranges as in normal aging, unless a family history was found. For all patients, a comprehensive clinical history was taken and neurological examinations were performed. All probands and sporadic patients were previously tested negative for the SLC20A2 by Sanger sequencing. We also recruited 200 healthy individuals with the same ethnicity as controls.
Mutation detection
We collected venous blood samples from all included individuals. Genomic DNA of probands and sporadic patients was extracted from the peripheral blood lymphocytes using the Qiamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). The entire exons and exon–intron boundaries of PDGFRB were amplified by PCR. Primer sequences to amplify all exons and exon–intron boundaries of PDGFRB are listed in Table 1. Sequences of the PCR products were determined with an ABI 3730XL automated DNA-sequencing system. The sequence data were analyzed and compared with reference sequences from the Human Genome database (NM_002609.3). Subsequently, the identified variants in probands or sporadic patients were screened in all available family members and 200 controls in order to evaluate whether the variant co-segregated with the disease in the family and whether it was present in the controls of matched geographical ancestry.
In silico analysis
We used Mutation Taster (http://www.mutationtaster.org), Polymorphism Phenotyping v2 (PolyPhen-2) (http://genetics.bwh.harvard.edu/pph2/) and Sorts Intolerant From Tolerant (http://sift.jcvi.org/www/SIFT_enst_submit.html) to predict the pathogenic potential of the identified variants on the function of the protein. Evolutionary conservation of the affected amino acid among different species is estimated by the HomoloGene (http://www.ncbi.nlm.nih.gov/homologene). We also assessed the novelty of the identified variants by searching the dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 Genomes Project (http://www.internationalgenome.org/home) and Exome Aggregation Consortium (ExAC) databases (http://exac.broadinstitute.org/).
Results
Direct sequencing of the PDGFRB gene revealed two heterozygous variants, c.3G>A and c.2209G>A, in two families, respectively (Figure 1). Both families were from South China. We further screened PDGFB and XPR1 in these two families and found no mutations. These two variants were not observed in 200 healthy controls of the same ethnicity. We did not find any PDGFRB mutations in included sporadic patients.

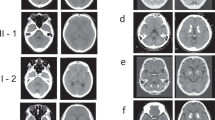
Pedigrees of two families with PFBC with PDGFRB mutations. (a) The pedigree and CT scans of Family 1; (b) the pedigree and CT scans of Family 2. The proband is marked by a black arrow; filled symbols indicate affected individuals; symbols with question marks indicate individuals with unknown status; symbols with a ‘+’ indicate mutation carriers and symbols with a ‘−’ indicate non-carriers; Calcification regions are indicated by white arrows.
The variant c.3G>A was detected in a two-generation family (Family 1, Figure 1a). Brain CT and blood samples were available in four members, including three affected individuals (I1, II2 and II3) and one unaffected individual (I2). The proband (II3) was a 38-year-old man who had no clinical symptoms (Table 2). Brain CT showed that the proband (II3, evaluated at 38 years) and his father (I1, evaluated at 67 years) had obvious calcification in bilateral globus pallidus and cerebellum, whereas his sister (II2, evaluated at 43 years) presented mild calcification in bilateral globus pallidus (Figure 1a). All of the affected individuals were clinical asymptomatic and carried the same variant (c.3G>A) in a heterozygous state (Figure 2a). The variant c.3G>A was located in exon 2 and affected the initiation codon of the PDGFRB gene. It resulted in a change from an ATG codon to an ATA codon and might replace the start methionine (p.M1?). Sorts Intolerant From Tolerant predicted this variant as damaging. Moreover, this variant was not found in the dbSNP, 1000 Genomes and ExAC databases.

Direct sequencing results and multiple sequence alignment analysis of PDGFRB mutations identified in this study. (a) Sequencing chromatograms of the two novel PDGFRB mutations; the identified mutations are indicated by red arrows. (b) Multiple alignment of protein sequence from different species; the mutated residue showing conservation is indicated by a box. A full color version of this figure is available at the Journal of Human Genetics journal online.
The variant c.2209 G>A was detected in a proband of a two-generation pedigree (Family 2, Figure 1b). We collected brain CT and blood samples from four members of this family (I1, I2, II8 and II9). The proband was a 45-year-old man having a 1-year history of dizziness (Table 2). Neurological examination showed no obvious abnormality. Brain CT of the proband (II8, evaluated at 45 years) and his mother (I2, evaluated at 67 years) showed mild calcification in bilateral globus pallidus, while his brother and his father had normal brain CT (Figure 1b). His mother was asymptomatic. The affected members (I2 and II8) in this pedigree were heterozygous for the variant c.2209 G>A, while the mutation was not detected in unaffected members (I1 and II9) (Figure 2a). These results suggested that this novel variant co-segregated with the disease phenotype in this family and may be a pathogenic variant causing PFBC. The variant c.2209 G>A resulted in amino-acid substitutions of aspartic acid to asparagine at position 737 (p.D737N). Mutation Taster and Polyphen-2 predicted this variant to be damaging. Furthermore, multiple alignment of amino-acid sequence revealed that the affected amino acid was highly conserved among the different species, suggesting its functional importance (Figure 2b). This missense variant was reported in ExAC databases at an extreme frequency of 0.000008 and absent in dbSNP and 1000 Genomes Project.
Discussion
Here we found two novel mutations in exons 2 and 16 of PDGFRB. Both of these two mutations co-segregated with the disease phenotype and was absent in any control individuals, indicating that they were not likely to represent a benign polymorphism. The mutation c.3G>A affected the initial codon leading to the elimination of initial methionine, and c.2209G>A, located in a highly conserved region, results in an amino-acid substitution that was predicted to be deleterious on protein function. According to the joint consensus recommendation for the interpretation of sequence variants in 2015,10 c.3G>A can be considered to be pathogenic (PVS1+PM2+PP1+PP4) and c.2209G>A can be considered as variant of unknown significance based on current evidence (PP1+PP2+PP3+PP4).
The PDGFRB gene encodes platelet-derived growth factor receptor type β (PDGFRβ), a cell surface tyrosine kinase receptor 11. PDGFRβ consists of an extracellular domain which binds the ligand (PDGFs), a transmembrane domain and an intracellular tyrosine-kinase domain (562-953aa).11 Binding of PDGFs to PDGFRβ leads to the autophosphorylation of PDGFRβ and then activate the downstream pathway. Previous studies have demonstrated that PDGFs/PDGFRβ signaling pathway has an important role in cell proliferation, migration, survival and differentiation.11, 12 Presently, identification of mutations in PDGFRB and its ligand PDGFB first reveals the link between PDGFB/PDGFRβ signaling and the development of brain calcifications. However, it remains unclear how impaired PDGFB/ PDGFRβ pathway contribute to the formation of brain calcification. The potential mechanisms seem to be mediated by the impairment of phosphorus homeostasis and the blood–brain barrier integrity.3, 5 To date, no loss-of-function mutation of PDGFRB has been reported to cause PFBC, whereas all PDGFB mutations lead to loss of a functional PDGFB protein.5, 9, 13, 14 Among the four reported mutations causing PFBC, p.R987W, p.L658P and p.R695C resulted in defective PDGFRB autophosphorylation, while p.E1071V, found in a sporadic case and absent in population database, was assumed to be represent a benign variant. This should draw our attention when classifying a rare variant as pathogenic. L658P and R695C locate in the protein kinase catalytic domain, whereas R987W and E1071V are outside of the tyrosine kinase domain. In our study, the mutation c.3G>A (p.M1?) replaced the start methionine and might result in a complete lack of PDGFRβ or a truncated protein that would lack the N-terminal signal peptide. Interestingly, we found an alternative ATG codon at position 7 (in frame) after checking the sequence of the PDGFRB gene. When this downstream ATG is used, six amino acids are removed from the N-terminal signal peptide, thereby decreasing its size from 1106 amino acids to 1100 amino acids. Given the short distance from original initiation codon, this variant might have a mild effect on the protein. However, the variant was still supported to be likely deleterious by bioinformatics analysis, segregation analysis and population data. Therefore, we still considered this variant as pathogenicity. The mutation c.2209 G>A (p.D737N) is located within the tyrosine kinase domain of PDGFRβ. The function studies have found that missense mutations in the tyrosine kinase domain may directly influence the activity of PDGFRβ and cause loss of function of the gene product.9, 14 We suppose that p.D737N mutation may also affect receptor activation and impair the PDGFB/ PDGFRβ pathway. Nevertheless, further study of the protein function is still needed to decipher the pathogenic mechanism of the variants identified in our study.
In our study, PFBC patients with PDGFRB mutations showed heterogeneous radiological phenotype and the majority of them were not clinically affected. Even patients carrying the same mutation in a family (Family 1) have obvious differences in the radiological phenotype, suggesting the wide phenotypic diversity. Nicolas et al.15 reported that PDGFRB mutation carriers seem to have a milder calcification compared with SLC20A2 and PDGFB mutation carriers. In our study, especially in Family 2, both the proband and his mother showed very mild calcification in the bilateral basal ganglia. Taking their old age into consideration, the brain calcification seemed to be physiological. Indeed, previous studies showed that brain calcification has also been reported in normal aging and seems to be common in older people. According to the scale of Nicolas et al.,15 calcification in Family 2 was rated below the age-specific threshold and the presence of calcifications in two relatives is not sufficient to classify it as PFBC. For this reason, two likely patients of Family 2 cannot be diagnosed with definite PFBC. However, we could not rule out the possibility of mild effect on calcification of the PDGFRB missense variant given the unknown significance of this variant. Therefore, we still need further functional study to confirm the pathogenicity of this variant and the diagnosis in this family.
Of note, patients with PFBC present clinical heterogeneity and many individuals with brain calcification can remain asymptomatic throughout life.2, 8, 16 A recent systematic review on the neuroimaging and clinical phenotype of PFBC showed that the penetrance of the clinical phenotype in PFBC is only 61% compared with 100% of the imaging phenotype.17 Given the gradually progressive symptoms of PFBC, we cannot exclude that they could develop disease manifestations with a long follow-up.
In conclusion, we found a pathogenic variant (c.3G>A) and a variant of unknown significance (c.2204G>A) for PFBC in Chinese patients. Further studies of the pathogenesis on these mutations might yield useful insights into the pathogenesis of PFBC. This is the first study to determine the prevalence of PDGFRB mutations in Chinese PFBC cohort. The prevalence of PDGFRB mutations in Chinese PFBC patients seems to be quite low, indicating that PDGFRB is not a common causal gene of PFBC in Chinese population.
References
Sobrido, M. J., Coppola, G., Oliveira, J., Hopfer, S. & Geschwind, D. H. in GeneReviews(R) (eds Pagon, R. A., Adam, M. P., Ardinger, H. H., Wallace, S. E., Amemiya, A., Bean, L. J. H. et al.) (Seattle, WA, USA, (1993).
Oliveira, J. R., Spiteri, E., Sobrido, M. J., Hopfer, S., Klepper, J., Voit, T. et al. Genetic heterogeneity in familial idiopathic basal ganglia calcification (Fahr disease). Neurology 63, 2165–2167 (2004).
Nicolas, G., Pottier, C., Maltete, D., Coutant, S., Rovelet-Lecrux, A., Legallic, S. et al. Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 80, 181–187 (2013).
Wang, C., Li, Y., Shi, L., Ren, J., Patti, M., Wang, T. et al. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat. Genet. 44, 254–256 (2012).
Keller, A., Westenberger, A., Sobrido, M. J., Garcia-Murias, M., Domingo, A., Sears, R. L. et al. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat. Genet. 45, 1077–1082 (2013).
Legati, A., Giovannini, D., Nicolas, G., Lopez-Sanchez, U., Quintans, B., Oliveira, J. R. et al. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat. Genet. 47, 579–581 (2015).
Lemos, R. R., Ramos, E. M., Legati, A., Nicolas, G., Jenkinson, E. M., Livingston, J. H. et al. Update and mutational analysis of SLC20A2: a major cause of primary familial brain calcification. Hum. Mutat. 36, 489–495 (2015).
Nicolas, G., Pottier, C., Charbonnier, C., Guyant-Marechal, L., Le Ber, I., Pariente, J. et al. Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification. Brain 136, 3395–3407 (2013).
Sanchez-Contreras, M., Baker, M. C., Finch, N. A., Nicholson, A., Wojtas, A., Wszolek, Z. K. et al. Genetic screening and functional characterization of PDGFRB mutations associated with basal ganglia calcification of unknown etiology. Hum. Mutat. 35, 964–971 (2014).
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Andrae, J., Gallini, R. & Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 22, 1276–1312 (2008).
Tallquist, M. D., Klinghoffer, R. A., Heuchel, R., Mueting-Nelsen, P. F., Corrin, P. D., Heldin, C. H. et al. Retention of PDGFR-beta function in mice in the absence of phosphatidylinositol 3'-kinase and phospholipase Cgamma signaling pathways. Genes Dev. 14, 3179–3190 (2000).
Vanlandewijck, M., Lebouvier, T., Andaloussi Mäe, M., Nahar, K., Hornemann, S., Kenkel, D. et al. Functional characterization of germline mutations in PDGFB and PDGFRB in primary familial brain calcification. PLoS ONE 10, e0143407 (2015).
Arts, F. A., Velghe, A. I., Stevens, M., Renauld, J. C., Essaghir, A. & Demoulin, J. B. Idiopathic basal ganglia calcification-associated PDGFRB mutations impair the receptor signalling. J. Cell Mol. Med. 19, 239–248 (2015).
Nicolas, G., Charbonnier, C., de Lemos, R. R., Richard, A. C., Guillin, O., Wallon, D. et al. Brain calcification process and phenotypes according to age and sex: Lessons from SLC20A2, PDGFB, and PDGFRB mutation carriers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 168, 586–594 (2015).
Wider, C., Dickson, D. W., Schweitzer, K. J., Broderick, D. F. & Wszolek, Z. K. Familial idiopathic basal ganglia calcification: a challenging clinical-pathological correlation. J. Neurol. 256, 839–842 (2009).
Tadic, V., Westenberger, A., Domingo, A., Alvarez-Fischer, D., Klein, C. & Kasten, M. Primary familial brain calcification with known gene mutations: a systematic review and challenges of phenotypic characterization. JAMA Neurol. 72, 460–467 (2015).
Acknowledgements
We sincerely thank the patients and their relatives for participation. This work was supported by the grant nos 81322017, 81371261 and U1505222 from the National Natural Science Foundation of China and grant NCET-13-0736 from the Program for New Century Excellent Talents in University, National Key Clinical Specialty Discipline Construction Program and Key Clinical Specialty Discipline Construction Program of Fujian.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Wang, C., Yao, XP., Chen, HT. et al. Novel mutations of PDGFRB cause primary familial brain calcification in Chinese families. J Hum Genet 62, 697–701 (2017). https://doi.org/10.1038/jhg.2017.25
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.25
This article is cited by
-
PDGF receptor mutations in human diseases
Cellular and Molecular Life Sciences (2021)
-
A novel PDGFRB sequence variant in a family with a mild form of primary familial brain calcification: a case report and a review of the literature
BMC Neurology (2019)
-
Basal ganglia calcifications (Fahr’s syndrome): related conditions and clinical features
Neurological Sciences (2019)
-
Primary brain calcification: an international study reporting novel variants and associated phenotypes
European Journal of Human Genetics (2018)
-
Clinical and radiological diversity in genetically confirmed primary familial brain calcification
Scientific Reports (2017)
