
Abstract
Cerebral, ocular, dental, auricular, skeletal (CODAS) syndrome is a rare autosomal recessive multisystem disorder caused by mutations in LONP1. It is characterized by intellectual disability, cataracts, delayed tooth eruption, malformed auricles and skeletal abnormalities. We performed whole-exome sequencing on a 12-year-old Japanese male with severe intellectual disability, congenital bilateral cataracts, spasticity, hypotonia with motor regression and progressive cerebellar atrophy with hyperintensity of the cerebellar cortex on T2-weighted images. We detected compound heterozygous mutation in LONP1. One allele contained a paternally inherited frameshift mutation (p.Ser100Glnfs*46). The other allele contained a maternally inherited missense mutation (p.Arg786Trp), which was predicted to be pathogenic by web-based prediction tools. The two mutations were not found in Exome Variant Server or our 575 in-house control exomes. Some features were not consistent with CODAS syndrome but overlapped with Marinesco–Sjögren syndrome, a multisystem disorder caused by a mutation in SIL1. An atypical mutation site may result in atypical presentation of the LONP1 mutation.
Similar content being viewed by others

Introduction
Cerebral, ocular, dental, auricular, skeletal (CODAS) syndrome (MIM 600373) is a rare autosomal recessive inherited multisystem disorder. It is characterized by developmental delay, cataracts, delayed tooth eruption, malformed auricles and skeletal abnormalities.1, 2 Its clinical spectrum ranges from typical cases with five major features to atypical cases with only two or three major features. CODAS syndrome phenotypes were recently found to be caused by mutations in LONP1, a gene encoding a mitochondrial adenosine triphosphate (ATP)-dependent protease.1, 2, 3
Marinesco–Sjögren syndrome (MSS; MIM 248800) is a rare autosomal recessive inherited multisystem disorder.4 The clinical triad consists of early onset bilateral cataracts, cerebellar ataxia and intellectual disability.5 Myopathy, motor regression, pyramidal signs, skeletal abnormalities and hypogonadism are seen at variable frequencies.6 Using brain magnetic resonance imaging (MRI), hyperintensity of the cerebellar cortex on T2-weighted images is the most specific and striking finding.7 Recently, mutations in SIL1, a gene encoding nucleotide exchange factors that function in the endoplasmic reticulum, were identified as the main cause of MSS.8 SIL1 mutations are detected in 60% of typical MSS cases but the mutation rate is lower than 3% in atypical MSS cases, suggesting that other genes are responsible for the atypical cases.4
Here we report a case of atypical CODAS syndrome with heterozygous mutations in LONP1. Early onset bilateral cataracts, intellectual disability, hypotonia with motor regression and progressive cerebellar atrophy with high intensity on MRI were consistent with MSS. This case reveals that the mutation of LONP1 can present similar features with that of SIL1.
Case Report
The proband is a 12-year-old male, born to healthy, non-consanguineous parents. He has one healthy brother. He was born by cesarean section due to placenta previa at 36 weeks of gestation. No asphyxia was recorded. His birth weight, length and head circumference were 2324 g (−0.6 s.d.), 42.6 cm (−1.5 s.d.) and 32.5 cm (+0.3 s.d.), respectively. Imperforate anus with rectovesical fistula and bilateral congenital cataracts were diagnosed at birth and were operated twice on at 1 day and 3 months, respectively.
He was referred to our hospital because of developmental delay at 10 months. Physical examination revealed short stature, hypotonia and spasticity of lower extremities. No facial dimorphism, including microcornea, auricular or teeth deformities, was seen. At 1 year 6 months, he could crawl but could not sit alone nor speak coherent words. At 2 years, he regressed and could not crawl. At 2 years 10 months, he presented with brief tonic seizures, which were controlled with valproate. Electric encephalogram showed rare spikes. At 8 years, a gastrostomy was placed for progressive swallowing difficulties. At 9 years, he presented with intermittent choreoathetotic movement.
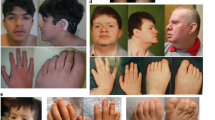
MRI performed at 10 months and 5 years showed progressive atrophy of the cerebellar cortex and caudate, with hyperintensity of the cerebellar cortex on T2-weighted images (Figures 1a–d). He had normal serum creatine kinase, amino acid, lactate, pyruvate, tandem mass spectroscopy and karyotype. Nerve conduction time and brainstem auditory evoked potentials were also normal. Skeletal radiography showed epiphyseal dysplasia on both knees (Figure 1e). Sanger sequencing denied SIL1 mutation.

(a–d) Brain MRI of the case. (a, b) Hyperintensity of the cerebellar cortex on T2-weighted images was visible at 10 months. (c,d) Progressive atrophy of cerebellar cortex and caudate and hyperintensity of the cerebellar cortex on T2-weighted images was seen at 5 years. (e) Skeletal radiography of the patient’s knees at 12 years. Epiphyseal dysplasia in both femoral metaphysis was seen.
Whole-exome sequencing identified compound heterozygous mutations in LONP1. One allele contained a paternally inherited frameshift mutation (NM_004793.3:c.296dup, p.(Ser100Glnfs*46)). The other allele contained a maternally inherited missense mutation (NM_004793.3:c.2356C>T, p.(Arg786Trp)), which was predicted to be pathogenic by web-based prediction tools (Supplementary Table S1). Arg786 is evolutionarily conserved from humans to insects in Lon protein sequences (Supplementary Figure S1). The two mutations segregated with the disease in the family, and were not found in Exome Variant Server (http://evs.gs.washington.edu/EVS/) or in our 575 in-house control exomes. The c.296dup mutation was not registered in ExAC database (http://exac.broadinstitute.org/), but the c.2356C>T was found in 9 of 119 692 alleles. No mutations were found in CTDP1.
Discussion
We report a patient with bilateral congenital cataracts, developmental delay, progressive cerebellar atrophy, hypotonia with motor regression and choreoathetotic movement. Because of the presence of the clinical triad, hypotonia with motor regression and characteristic MRI findings, he was suspected of having MSS. Mutations in SIL1 were excluded by Sanger sequencing and whole-exome sequencing. Other differential diagnoses, including congenital cataracts, facial dysmorphism and neuropathy, were ruled out based on the clinical and genetic features.
In this case, we detected mutations in LONP1. Auricle malformation and delayed tooth eruption were not present. Regression, involuntary movement and cerebellar atrophy with high intensity on MRI have not been reported in CODAS syndrome, so this case was not consistent with typical CODAS syndrome.
LONP1 codes for Lon protease, a multi-functional mitochondrial enzyme with diverse roles, including (1) elimination of misfolded and oxidatively damaged proteins, (2) chaperone-like assembly of protein complexes within the respiratory chain and (3) regulation of mitochondrial gene expression.1 Various allele combinations that result in alteration of LON function are thought to cause variations in the presentation of symptoms.1 In typical CODAS syndrome with five major features, pathogenic mutations cluster near the ATPase domain, but in atypical CODAS syndrome without five major features, the mutations spread to other domains.1, 2, 3 In this case, one mutation was a missense mutation in the proteolytic domain, and the other was a frameshift mutation in the N-terminal domain (Figure 2), which may result in atypical presentation of the LONP1 mutation. So far, some LONP1 mutations in the ATPase domain are reported to result in reduced enzyme activity. There is no report about correlation between mutations other than in the ATPase domain and enzyme activity. Further studies are needed to confirm how mutations besides ATPase domain affect biochemically.

Functional domains of the human LON subunit. Reported mutations and the mutation identified in this case are shown. Missense mutations are shown above the schematic; frameshift and nonsense mutations are shown below. The black circles indicate the mutations of typical CODAS syndrome, the white triangles indicate the mutations of atypical CODAS syndrome and the arrow indicates the mutations of this case. Each circle, triangle, and arrow represents each mutation. Mitochondrial targeting sequence, N-terminal domain, ATPase domain and a proteolytic domain. A full color version of this figure is available at the Journal of Human Genetics journal online.
The intellectual disability, cerebellar atrophy, hypotonia and regression were common to other mitochondrial diseases.9 Congenital cataracts and movement disorders are sometimes seen in other mitochondrial disorders. It is plausible that dysfunction of LON, a mitochondrial chaperone enzyme, results in mitochondrial dysfunction and could show similar characteristics.
Some features in this case were not consistent with CODAS syndrome but overlapped with MSS, including cerebellar atrophy with high intensity in MRI, regression and spasticity. Most MSS cases are caused by a mutation in SIL1, a gene encoding nucleotide exchange factors of the endoplasmic reticulum. Mitochondria and endoplasmic reticulum have multiple contact sites forming specific domains, and dysfunction of SIL1 causes decreased levels of many mitochondrial membranes and membrane-binding proteins.10 In fact, the mitochondrial architecture is altered in Sil1-depleted mice.11 In addition, one of the SIL1 mutation-negative atypical MSS patients was associated with a mitochondrial DNA depletion disorder caused by AGK mutation.4 AGK encodes acylglycerol kinase, a mitochondrial membrane protein that acts as a multi-substrate lipid kinase.12 Moreover, AGK expression is decreased by SIL1 depletion.10 In this case, LONP1 mutations probably caused some features consistent with MSS due to mitochondrial dysfunction. This is the first case showing that a LONP1 mutation overlaps with some features of MSS. This case also suggests a correlation between MSS and mitochondrial dysfunction, although more cases are required for confirmation.
In conclusion, we show that LONP1 mutations exhibit atypical presentation with CODAS syndrome. Some features are consistent with MSS, which may be caused by mitochondrial dysfunction.
References
Strauss, K. A., Jinks, R. N., Puffenberger, E. G., Venkatesh, S., Singh, K., Cheng, I. et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 96, 121–135 (2015).
Dikoglu, E., Alfaiz, A., Gorna, M., Bertola, D., Chae, J. H., Cho, T. J. et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. A 167, 1501–1509 (2015).
Venkatesh, S., Lee, J., Singh, K., Lee, I. & Suzuki, C. K. Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim. Biophys. Acta. 1823, 56–66 (2012).
Krieger, M., Roos, A., Stendel, C., Claeys, K. G., Sonmez, F. M., Baudis, M. et al. SIL1 mutations and clinical spectrum in patients with Marinesco-Sjogren syndrome. Brain 136, 3634–3644 (2013).
Sjögren, T. Hereditary congenital spinocerebellar ataxia accompanied by congenital cataract and oligophrenia; a genetic and clinical investigation. Confin. Neurol. 10, 293–308 (1950).
Goto, M., Okada, M., Komaki, H., Sugai, K., Sasaki, M., Noguchi, S. et al. A nationwide survey on Marinesco-Sjögren syndrome in Japan. Orphanet. J. Rare Dis. 9, 58–66 (2014).
Harting, I., Blaschek, A., Wolf, N. I., Seitz, A., Haupt, M., Goebel, H. H. et al. T2-hyperintense cerebellar cortex in Marinesco-Sjögren syndrome. Neurology 63, 2448–2449 (2004).
Senderek, J., Krieger, M., Stendel, C., Bergmann, C., Moser, M., Breitbach-Faller, N. et al. Mutations in SIL1 cause Marinesco-Sjögren syndrome, a cerebellar ataxia with cataract and myopathy. Nat. Genet. 37, 1312–1314 (2005).
DiMauro, S., Schon, E. A., Carelli, V. & Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 9, 429–444 (2013).
Roos, A., Kollipara, L., Buchkremer, S., Labisch, T., Brauers, E., Gatz, C. et al. Cellular signature of SIL1 depletion: disease pathogenesis due to alterations in protein composition beyond the ER machinery. Mol. Neurobiol. 53, 5527–5541 (2016).
Roos, A., Buchkremer, S., Kollipara, L., Labisch, T., Gatz, C., Zitzelsberger, M. et al. Myopathy in Marinesco-Sjögren syndrome links endoplasmic reticulum chaperone dysfunction to nuclear envelope pathology. Acta Neuropathol. 127, 761–777 (2014).
Mayr, J. A., Haack, T. B., Graf, E., Zimmermann, F. A., Wieland, T., Haberberger, B. et al. Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am. J. Hum. Genet. 90, 314–320 (2012).
Acknowledgements
We thank the patient and the patient’s family for the participation in this study. This study was supported partly by Intramural Research Grant (26-8) for Neurological and Psychiatric Disorders of NCNP and Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and Development, AMED (IN), a Grant-in-Aid for Scientific Research (B) (25293085) (HS), Grants from Research on Measures for Intractable Diseases (NM), Comprehensive Research on Disability Health and Welfare (NM), the Strategic Research Program for Brain Science (NM), the Initiative on Rare and Undiagnosed Diseases in Pediatrics (NM), the Initiative on Rare and Undiagnosed Diseases for Adults (NM) from the Japanese Agency for Medical Research and Development; a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the fund for the Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems (NM) from the Japanese Science and Technology Agency; the Takeda Science Foundation (NM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
IN received honoraria from Sanofi and research funding from Astellas Pharma although there is no conflict with this study. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Inui, T., Anzai, M., Takezawa, Y. et al. A novel mutation in the proteolytic domain of LONP1 causes atypical CODAS syndrome. J Hum Genet 62, 653–655 (2017). https://doi.org/10.1038/jhg.2017.11
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.11
This article is cited by
-
Structures of the human LONP1 protease reveal regulatory steps involved in protease activation
Nature Communications (2021)
-
Mitochondrial Lon protease is a gatekeeper for proteins newly imported into the matrix
Communications Biology (2021)

{kind=link}