
Abstract
With the advance of next-generation sequencing technology, the rare variants join the common ones in explaining more proportions of heritability. The coexistence of variants of common with rare, causal with neutral and deleterious with protective is a norm and should be appropriately addressed. Some existing methods suffer from low power when one or more forms of coexistence present, impeding their applications in practice. In this paper, for case–parent trios, pseudocontrols are constructed using the nontransmitted alleles of the parents. The Kullback–Leibler divergence is utilized to measure the difference between the distributions of variants in a genetic region for the affected children and pseudocontrols, and two nonparametric test statistics KLTT and cKLTT are proposed. Extensive simulations show that they are robust to the opposite directions of the causal variants and the amount of neutral variants, and have superiority over the existing methods when both rare and common variants are involved. Furthermore, their efficiency is demonstrated in the application to the data from Framingham Heart Study.
Similar content being viewed by others


Introduction
Benefitted from the Human Genome Project, the genome-wide association studies have identified hundreds of associated common variants (minor allele frequency (MAF) ⩾1%) under the common disease–common variant assumption.1 However, these variants explain only 5–10% of the disease burden in the population.2, 3 To uncover the missing heritability, the common disease–rare variant (MAF <1%) assumption was proposed.4, 5 With the advent of next-generation sequencing, many rare variants are detected to explain the missing heritability, such as obesity and hypertension.6, 7 The substantial evidence shows that both the common disease–common variant and the common disease–rare variant assumptions are valid, and the susceptibility genes probably involve the functional variants that range from rare to common.8 Furthermore, the functional genetic variants may have opposite effects (deleterious and protective).9, 10
The population- and family-based studies, having their own advantages and disadvantages, are two main forms in genome-wide association studies. Because of the intrinsic ease of collecting large data sets, the former has wider popularity than the latter.11 However, family-based study has unique advantages, as it is robust against population admixture and stratification, is able to identify technological artifacts in the data and has potential to detect more susceptibility loci.12 Furthermore, family-based study containing both within- and between-family information has substantial benefits in terms of multiple hypothesis testing.13 For the population-based study, cases and controls are chosen randomly from affected and unaffected populations, and all involved subjects are then independent. Single-marker test is a primary approach to detect common variants; meanwhile, some efficient and powerful methods such as the sequence kernel association test14 and the adaptive sum of powered score test15 have been proposed accordingly for rare variants association study.
For family data, the transmission/disequilibrium test (TDT)16 and family-based association test (FBAT)17 are two classic association methods. De et al.18 collapsed the standard statistic of FBAT in a genetic region and developed the test statistic specially for rare variants. Ionita-Laza et al.19 proposed the family-based sequence kernel association test for the family data that is parametric and needs not only families with affected children, but also families with unaffected children. He et al.20 incorporated combined multivariate and collapsing (CMC),21 burden of rare variants test (BRT)22 and weighted sum statistic (WSS)23 into the TDT framework and proposed the corresponding ones. Based on sum of squared score test (SSU)24 and TDT, Preston and Dudbridge25 developed score statistics for the trios, where the haplotype phases were derived using BEAGLE.26 However, the accuracy of phasing with BEAGLE has upper limit because of the incomplete information contained in a given data set. In addition, Zhu and Xiong27 incorporated the matrix of kinship coefficients into CMC,21 and general T2 test to detect rare variants based on the family data. Sha and Zhang28 and Choi et al.29 constructed the conditional likelihood function of affected offspring given parents or siblings, and proposed the likelihood ratio test to detect rare variants. However, these methods would be vulnerable when some variants are deleterious to the disease whereas some variants are protective. Meanwhile, some existing methods are only applicable to rare variants, and thus exclude commons variants. Hence, it is necessary to develop methods handling common and rare variants simultaneously.
Like TDT, our test statistics are developed for the standard trios (father, mother and an affected child). We utilize the Kullback–Leibler divergence30, 31 to measure the distributional difference of the transmitted alleles and the nontransmitted alleles to the affected offspring from parents across a genetic region harboring common and rare variants with opposite effects, forming our first test statistic. Its derivate is introduced based on the comparison of the counts of transmitted and nontransmitted alleles at each site in this region. We design extensive simulation settings to assess empirically the performance of the proposed test statistics, where various levels of linkage disequilibrium (LD) among variants are addressed. Meanwhile, we compare them with some existing methods, of which some need to infer the phase. The results show that the proposed methods are almost more powerful than the existing methods in a range of scenarios, and are recommended in the presence of both common and rare variants with opposite effects. Finally, we apply the proposed methods to analyze the Framingham Heart Study (FHS) data. Several significant genes are detected, and most of them have been reported in the literature. The gene function enrichment analyses via g:Profiler (http://biit.cs.ut.ee/gprofiler/) further verify that these significant genes have some associations with the hypertension.
Materials and methods
Assume there are m sites in a genetic region where both common and rare variants may be present. For n case–parent trios, let F=(Fij)n × m, M=(Mij)n × m and C=(Cij)n × m denote the genotype matrices for the fathers, mothers and affected children, respectively, where Fij (Mij, Cij) being 0, 1 or 2 is the copy number of minor allele at the jth site for the father (mother, child) in the ith trio, respectively, i=1,…, n and j=1,…, m.
For every given case–parent trio and at each site, as one knows both parents have one allele that is not transmitted to the affected child, these two nontransmitted alleles could be combined to construct a pseudocontrol. As nontransmitted alleles serve as controls that have the same population genetic background as the affected children, more findings are anticipated from the genotype comparison of affected children and pseudocontrols. Based on above-mentioned allele coding scheme, Fij+Mij−Cij is actually the copy number of the minor allele of the pseudocontrol at site j in the ith trio, i=1,…, n and j=1,…, m. It is so plausible to regard Fij+Mij−Cij as the genotype of the pseudocontrol. Consequently, we have a group of pseudocontrols having the same number as that of the affected children.
It is shown in the Supplementary Information that under the null hypothesis of no association, Cij is independent of Fij+Mij−Cij for the same locus j (Supplementary Table S1), and Cik is independent of Fij+Mij−Cij for highly linked loci k and j (Supplemenetary Table S2). Hence, C is independent of F+M−C under the null hypothesis. That is to say, genetic variants are present randomly in the region of m sites for both affected children and pseudocontrols. Thus, the distribution of genetic variants for affected children is roughly the same as that for pseudocontrols. Some difference between the genotypes of these two groups would be expected if the disease is associated with one or more variants. In the following, we utilize the Kullback–Leibler divergence30, 31 to measure the difference between distributions of variants for the affected children and pseudocontrols that forms our primary test statistic.
Let aj and bj denote the copy number of the minor allele at site j of the affected children and pseudocontrols across n trios, j=1,…, m, respectively; that is,

Then we calculate the relative frequency of variant at site j among all m sites for the respective affected children and pseudocontrols as follows,

where the constant 1 is added to the counts to ensure fj>0 and gi>0 for every j. It is natural to regard f={f1,…, fm} and g={g1,…, gm} as the distributions of genetic variants for the affected children and pseudocontrols, respectively. For the probability mass functions f and g having the same support, we calculate the Kullback–Leibler divergence between f and g as

Similarly, we compute the Kullback–Leibler divergence between g and f as  .
.
Note that neither H(f, g) nor H(g, f) is symmetric about f and g. In order to construct a symmetric measure of difference between f and g, we adopt the following form:

that is our first test statistic, Kullback–Leibler divergence-based Test for Trios. It is time to investigate some property of KLTT based on its form. As people have already realized, some genetic variants may be deleterious to diseases whereas some others may be protective. Roughly speaking, we could imply fj>gj for the deleterious variant at site j and fj<gj for the protective one that always lead to a positive summand (fj−gj)(log fj−log gj) in the formula of KLTT. It is so anticipated that KLTT has the potential to efficiently detect the variants of positive and negative associations simultaneously. It is also noted from the sum expression in Equation (1) that KLTT considers common and rare variants together without the worry of contribution from one type overshadowing the other.
In addition, we also build the test statistic using copy numbers  and
and  instead of
instead of  and
and  in the expression of KLTT. The corresponding test statistic is
in the expression of KLTT. The corresponding test statistic is

where the constant 1 is added to the counts to prevent 0 in the log operation. It is observed from Equations (1) and (2) that KLTT measures the difference between relative frequencies  and
and  whereas cKLTT measures the difference between frequencies
whereas cKLTT measures the difference between frequencies  and
and  .
.
In order to assess the performances of KLTT and cKLTT thoroughly, we need to compare them with the existing methods in He et al.,20 Preston and Dudbridge25 and Choi et al.29 in terms of detection power. Hence, we give a brief description of these methods for ease of reference. He et al.20 incorporated rare-variant association methods CMC,21 BRT22 and WSS23 into the TDT16 framework, where the phasing was performed with BEAGLE.26 Let clj=1 (dlj=1) if a minor-allele (major-allele) transmitted event occurs for parent l with variant j, otherwise 0, l=1,…, 2n, j=1,…, m. They then constructed the counterparts of b and c in the 2 × 2 table of TDT based on CMC, BRT and WSS, and adopted the form of TDT test (b−c)2/(b+c)16 to detect genetic variants. He et al.20 indicated TDT-WSS performed well in most scenarios. Hence, in this paper we compare our methods with TDT-WSS, in which  and
and  , where ωj is the estimated s.d. of the MAF at locus j based on all pseudocontrols. Moreover, to be simple, we generate haplotype data and the phases are known in simulation studies.
, where ωj is the estimated s.d. of the MAF at locus j based on all pseudocontrols. Moreover, to be simple, we generate haplotype data and the phases are known in simulation studies.
Preston and Dudbridge25 devised five new family-based score statistics based on Pan.24 Let E={Eij}n × m (H={Hij}n × m) denote the count of minor (major) alleles transmitted to the affected offspring from the parents who are heterozygous at the jth variant for the ith trio, i=1,…, n, j=1,…, m. Score vector is defined as U=XT1 and its variance–covariance matrix is then  , where X=(1/2)(E−H),
, where X=(1/2)(E−H),  ,
,  with
with  , and 1 is the all 1 vector of length n. As done in Pan,24 the family-based score statistics are proposed accordingly, denoted as Tscore, TSSU, TSSUw, TUminP and Tsum. To save the space, TSSU=UTU, that was demonstrated to have the outstanding performance among them,25 is selected to compare with our proposed ones.
, and 1 is the all 1 vector of length n. As done in Pan,24 the family-based score statistics are proposed accordingly, denoted as Tscore, TSSU, TSSUw, TUminP and Tsum. To save the space, TSSU=UTU, that was demonstrated to have the outstanding performance among them,25 is selected to compare with our proposed ones.
Choi et al.29 proposed a FAmily-based Rare Variant Association Test (FARVAT) based on the quasilikelihood of whole families. Let  and Yi be the genotype and phenotype (0=unaffected; 1=affected) vectors in a family i for variants j, respectively, and
and Yi be the genotype and phenotype (0=unaffected; 1=affected) vectors in a family i for variants j, respectively, and  , and
, and  , i=1,…, n, j=1,…, m. The score tests for the C-alpha-type test (FARVATc) and the burden-type test (FARVATb) were devised from
, i=1,…, n, j=1,…, m. The score tests for the C-alpha-type test (FARVATc) and the burden-type test (FARVATb) were devised from  and Var(Pj|Y)=σjjΦ, where pj is the MAF of variant j; N is the total number of individuals in n families; 1N is the N × 1 column vector that consisted of 1; and Φ denotes the kinship matrix. FARVATb has an apparent weakness and its performance deteriorates much when causal variants have the opposite directions. Thus, FARVATc is chosen for the comparison.
and Var(Pj|Y)=σjjΦ, where pj is the MAF of variant j; N is the total number of individuals in n families; 1N is the N × 1 column vector that consisted of 1; and Φ denotes the kinship matrix. FARVATb has an apparent weakness and its performance deteriorates much when causal variants have the opposite directions. Thus, FARVATc is chosen for the comparison.
For our proposed test statistic T (KLTT or cKLTT), the permutation procedure is employed to evaluate its P-value. More specifically, the multisite genotypes of the affected child and the pseudocontrol are exchanged with probability 0.5 within each trio. This procedure is repeated B times, and we obtain the corresponding test statistic Tb for b=1,2,…, B. The P-value of the test statistic is given as

where I is the indicator function. Let Pr be the P-values, r=1,2,…, R, for R replications, the power (or type I error rate) for a given significance level α is calculated as

Results
Simulation study
Simulation setting
Extensive simulation settings are designed to evaluate the performance of KLTT and cKLTT, and to compare them with some existing methods.20, 25, 29 To generate the genotypes of trios, we first generate parents’ multisite genotypes based on a multivariate normal distribution. To be specific, we generate a latent vector Z=(Z1,…, Zm)T from a multivariate normal distribution with mean E(Zi)=0, variance Var(Zi)=1, i=1,…, m, and covariance described below. As we know, there may exist LD among genetic variants. To take this into account, we adopt the AR(1) model and set the correlation to be Corr(Zi,Zj)=ρ|i−j| if variants i and j are both causal or both noncausal, otherwise the correlation is 0. We set ρ=0, 0.5 and 0.9 to represent, to some extent, the no, moderate and strong LD, respectively. Each Zi is then transformed to 0 (major allele) or 1 (minor allele) determined by the corresponding MAF. The details for generating MAFs are given in the following section. This process repeats twice, and two 0-1 vectors of length m are put together to form the genotype of a parent. Once we have the genotypes of both parents, we then generate child’s genotypes under Mendelian inheritance. Note the recombination fraction between any two sites is 0 in this framework. The following logistic regression model is used to determine the disease status D of the child:

where β0 represents the logit of phenocopy rate or background disease prevalence, Gj is the genotype of the child at site j, ORj is the odds ratio of the jth genetic variant that represents its size of effect on the disease. In our simulation study we set m=32 and β0=log(0.1), corresponding to ∼9% phenocopy rate.
Table 1 shows the diversity of parameter settings. The MAFs of rare variants (causal or noncausal) are randomly generated from the uniform distribution U (0.001, 0.01); meanwhile, the MAFs of common variants are from the uniform distribution U (0.01, 0.5). To investigate the effect of different proportions of causal variants, different proportions of rare variants, different proportions of causal rare variants in causal variants and different proportions of positive effect sizes on the statistical power of tests, we design a total of 62 combinations of the numbers of causal rare variants, causal common variants (CCVs), noncausal rare variants and non-CCVs. See details in Table 1, where the proportion of causal (rare and common) variants in all 32 variants is 12.5, 25, 37.5 and 50%, and the proportion of rare (causal or noncausal) variants is 100% (or 30/32 × 100%, or 28/32 × 100%), 75, 50 and 25% (or 12/32 × 100%). For a given number of causal variants, we let all causal ones are rare or a big part are rare. For example, for the case of 16 causal variants (corresponding to 50% causal at the bottom in Table 1), we let the ratio of the number of causal rare variants to that of causal common ones be 16:0 and 12:4, as shown in Table 1. For each of these two ratios, we consider two types of the effect sizes; that is, log odds ratios. The first one is that exactly 50% effect sizes are positive and the sum of all effect sizes is 0. The other is that the number of positive effect sizes is bigger than the number of negative and the sum of all effect sizes is positive. Based on these parameter settings, we thus can evaluate comprehensively the performance of our proposed and existing tests.
To evaluate the type I error rate, we alter all odds ratios in Table 1 as 1. In the simulation study, we generate 400 trios, and the numerical results of powers of KLTT, cKLTT, TDT-WSS and TSSU are calculated via permutation procedure. The empirical powers/the type I error rates are evaluated based on 1000 replications and 200 permutations. The nominal significance level is set as 0.05. FAVRATc in Choi et al.29 follows the mixed χ2 distribution, and its significance is calculated with the Davies method.32
Simulation results
We first show the type I error rates of our proposed two test statistics and three existing ones20, 25, 29 with various LD structures in Supplementary Tables S3–S5 in the Supplementary Information. It is observed that all empirical sizes are around the significance level 0.05, and are well under control.
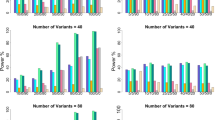
The statistical powers of five tests with no CCVs are depicted in Figure 1 (the sum of all effect sizes is 0) and Figure 2 (the sum of all effect sizes is positive), with 2 or 4 CCVs in Figure 3 (the sum of all effect sizes is 0) and Figure 4 (the sum of all effect sizes is positive). We could make some comments based on these results. First, for the situation in which the sum of log odds ratios is 0 (Figures 1 and 3), KLTT and cKLTT have almost the same powers and are the most powerful when both rare and common variants are involved. That is to say, when the candidate genetic region harbors both (causal or noncausal) rare and common variants, both deleterious and protective variants, the tests KLTT and cKLTT could detect functional variants powerfully. It is observed from Figure 1 that the powers of KLTT and cKLTT have a more than 10% increase compared with that of FARVATc in Choi et al.,29 are almost more than 1.5 times the powers of TDT-WSS in He et al.,20 and more than 2 times the powers of TSSU in Preston and Dudbridge,25 respectively. For example, in the situation of 12.5% causal and 25% rare with no LD (ρ=0), the powers of KLTT and cKLTT are 43.0%, and the powers of FARVATc, TDT-WSS, and TSSU are 37.7%, 14.6% and 7.3%, respectively (see Figure 1). The similar conclusions could be drawn when the number of CCVs equals 2 or 4 (see Figure 3). KLTT and cKLTT have the parallel superiority comparing with FARVATc, TDT-WSS and TSSU in the situation in which the sum of log odds ratios is positive (see Figures 2 and 4).

Powers of KLTT, cKLTT, TSSU, TDT-WSS and FARVATc against LD amount when the sum of all effect sizes is 0. Each of the 15 subfigures represents a combination of numbers of causal rare variants, noncausal rare variants and non-CCVs; the total number of variants is 32 and the number of CCVs is 0. The proportions of causal variants in the four row blocks are 12.5, 25, 37.5 and 50%, in the order from top to bottom, and the proportions of rare variants in the four column blocks are 25, 50, 75 and 100%, in the order from left to right. CCV, causal common variant; FARVAT, family-based rare variant association test; LD, linkage disequilibrium; SSU, sum of squared score; TDT, transmission/disequilibrium test; WSS, weighted sum statistic.

Powers of KLTT, cKLTT, TSSU, TDT-WSS and FARVATc against LD amount when the sum of all effect sizes is positive. Each of the 15 subfigures represents a combination of numbers of causal rare variants, noncausal rare variants and non-CCVs; the total number of variants is 32 and the number of CCVs is 0. The proportions of causal variants in the four row blocks are 12.5, 25, 37.5 and 50%, in the order from top to bottom, and the proportions of rare variants in the four column blocks are 25, 50, 75 and 100%, in the order from left to right. CCV, causal common variant; FARVAT, family-based rare variant association test; LD, linkage disequilibrium; SSU, sum of squared score; TDT, transmission/disequilibrium test; WSS, weighted sum statistic.

Powers of KLTT, cKLTT, TSSU, TDT-WSS and FARVATc against LD amount when the sum of all effect sizes is 0. Each of the 16 subfigures represents a combination of numbers of causal rare variants, noncausal rare variants and non-CCVs; the total number of variants is 32 and the number of CCVs is respectively 2 and 4 in the first and last two row blocks, in the order from top to bottom. The proportions of causal variants in the four row blocks are 12.5, 25, 37.5 and 50%, in the order from top to bottom; the proportion of rare variants is 50% and 75% in the second and third column blocks as indicated; is respectively 25% and 12/32 × 100% in the top three subfigures and the bottom one within the first column block; and is respectively 30/32 × 100% and 28/32 × 100% in the top two subfigures and bottom two ones within the last column block. CCV, causal common variant; FARVAT, family-based rare variant association test; LD, linkage disequilibrium; SSU, sum of squared score; TDT, transmission/disequilibrium test; WSS, weighted sum statistic.

Powers of KLTT, cKLTT, TSSU, TDT-WSS and FARVATc against LD amount when the sum of all effect sizes is positive. Each of the 16 subfigures represents a combination of numbers of causal rare variants, noncausal rare variants and non-CCVs; the total number of variants is 32 and the number of CCVs is respectively 2 and 4 in the first and last two row blocks, in the order from top to bottom. The proportions of causal variants in the four row blocks are 12.5, 25, 37.5 and 50%, in the order from top to bottom; the proportion of rare variants is 50% and 75% in the second and third column blocks as indicated; is respectively 25% and 12/32 × 100% in the top three subfigures and the bottom one within the first column block; and is respectively 30/32 × 100% and 28/32 × 100% in the top two subfigures and bottom two ones within the last column block. CCV, causal common variant; FARVAT, family-based rare variant association test; LD, linkage disequilibrium; SSU, sum of squared score; TDT, transmission/disequilibrium test; WSS, weighted sum statistic.
Second, the superiority of KLTT over cKLTT is exhibited when the LD level is strong (ρ=0.9) and the sum of log odds ratios is positive (see Figures 2 and 4). The smaller the number of rare variants or causal variants is, the more superiority it exhibits. For instance, the power of KLTT in the situation of 12.5% causal and 25% rare in Figure 2 with ρ=0.9 is 51.0%, whereas it is 34.7% for cKLTT. Third, the powers of TSSU and TDT-WSS are surprisingly low for scenarios in which both rare and common variants are involved. This may be partially because these two methods do not distinguish between common variants and rare variants and assign them the same weights in the test statistics. Finally, the ratio of the number of noncausal rare variants to that of noncausal common ones almost does not affect the powers of KLTT and cKLTT when the proportion of causal variants is fixed (see each row block of Figures 1,2,3,4). Meanwhile, the powers of FARVATc, TDT-WSS and TSSU decrease when the proportion of rare variants decreases, especially TSSU. Note that TSSU is a sum of squared score; that is, the difference between the counts of transmitted minor alleles and nontransmitted ones that suffers from substantial loss of power when both rare and common variants are present.
In scenarios involving only rare variants (100% rare, Figures 1 and 2), the winner goes to TSSU, followed by our proposed methods. To be desirable, the gap between our proposed methods and the most powerful method narrows with the increasement of LD amount. For example, in situation of 12.5% causal (Figure 1) with no LD, the powers of KLTT and TSSU are 40.7% and 60.2%, respectively, whereas with strong LD they are 13.8% and 19.4%, respectively. Fortunately, these scenarios of 100% rare are not norms in practice. Common diseases, not like Mendelian diseases, are usually associated with many genetic variants whose MAFs range from rare to common, even many genes. Moreover, cKLTT is superior to KLTT in situation in which all noncausal variants are rare (see the last column blocks in Figures 1,2,3,4) that may be partially explained as follows. cKLTT measures the difference between frequencies of copy numbers for the affected children and pseudocontrols directly, and this is more sensitive than the relative frequencies measured by KLTT.
It is also observed from Figures 1,2,3,4 that the LD level could affect the powers of all testing methods. On the one hand, the increased amount of LD between genetic variants with opposite effect directions (see Figures 1 and 3) could reduce the powers of all test statistics. For example, the powers of KLTT, cKLTT, FARVATc, TDT-WSS and TSSU in the situation of 50% rare and 50% causal with no LD (ρ=0) are 83.3%, 83.9%, 81.8%, 34.1% and 7.7%, respectively, versus 19.9%, 17.1%, 10.3%, 12.2% and 6.3% respectively, with strong LD (see Figure 1). Nevertheless, KLTT and cKLTT are still more powerful than the existing three test statistics in these scenarios. To give a direct interpretation, let us mimic two genetic variants in perfect LD having opposite effect directions with the same absolute value of effect sizes, and then their collective effect would become weak. Notice the kernel part of TDT-WSS is the difference of two sums that perhaps implies that their methods have low power to detect these two genetic variants. Whereas the distributions of relative frequencies or copy numbers of minor alleles for the affected children and pseudocontrols are different, our proposed methods still have the deserved power. On the other hand, the strong LD between genetic variants could increase the powers in situation of 50% causal in Figure 4, where the number of CCVs is 4 and the proportion of deleterious variants in all causal ones is more than one half, the powers of 5 methods are increasing when ρ is from 0.5 to 0.9. For example, the power of KLTT in Figure 4 with 50% causal and 75% rare is 59.2% (ρ=0.5) versus 79.6% (ρ=0.9).
Real data analysis
In this section, we use the proposed methods to analyze FHS data. FHS data are made available through the database of Genotypes and Phenotypes (dbGap)33 supplied by the Genetic Analysis Workshop 16. FHS participants are readily divided into three groups: the original cohort, the offspring cohort and the third-generation cohort, consisting of 5209, 5124 and 4095 participants, respectively. FHS data contain 1538 families whose mean pedigree size is 10 and ranges from 3 to 639. Owing to the existence of missing data, only 6849 participants have genotype data at 48 060 single-nucleotide polymorphism markers over the 22 autosomes.
FHS data contain systolic blood pressure, diastolic blood pressure, high-density lipoprotein cholesterol and other phenotypes. Here we focus on hypertension that results from a complex interaction of genes and environmental factors. Hypertension is usually defined as blood pressure ⩾140 mm Hg systolic or ⩾90 mm Hg diastolic blood pressure. As a prospective cohort study, FHS shows that the phenotype of the original cohort and the offspring cohort are measured in four examinations, whereas the third generation are only measured in one examination. Therefore, each participant is classified as either affected hypertension or not based on his/her highest measurement among all available systolic or diastolic blood pressure to minimize medication effect.
As KLTT (the results of simulation show that cKLTT is almost the same as KLTT, we hence drop cKLTT here) is used to analyze trio data, we first select affected participants and their parents, and then exclude families with missing mothers or fathers. Notice that we select only one trio from each pedigree to guarantee that all trios for our analysis are independent. In all, 113 trios are involved in the analysis. In this practice, we analyze all variants simultaneously in each gene. If FHS data provide only one variant for a gene, we combine 9 variants in its vicinity to form a region for analysis. The total number of genes is 14 067. MAFs of all single-nucleotide polymorphisms in the genes range from 0.0022 to 0.5 and the proportion of rare variants is 3.8% (Supplementary Figure S1). Note that we exclude TDT-WSS in He et al.20 as it needs the phase of every subject, which is not available in FHS genotype data. To evaluate the significance of each gene, we adopt 103 permutations. If the P-value is <10−3, we increase the times of permutation to 106.
Table 2 provides a summary of the top 10 significant results. Based on literature review, we learn that most of these 10 significant genes have been investigated in studies related to hypertension. For example, SORBS1 genetic variations contribute to insulin resistance, obesity, type 2 diabetes and hypertension.34 Gene EIF2AK1 is located in chromosome 7p22 whose mutations cause the familial hyperaldosteronism type II based on linkage analysis.35, 36 Familial hyperaldosteronism type II is an inherited form of hyperaldosteronism associated with hypertension in most patients. The ACSM3 gene, located on chromosome 16p12-13, encodes for enzymes catalyzing the activation of medium chain length fatty acids. Association studies have linked it to traits of insulin resistance syndrome and hypertension.37, 38 Sharma et al.39 suggested that the 20-ketosteroid reductase activity of the human AKR1C3 isozyme inactivates deoxycorticosterone that binds to the mineralocorticocoid receptor with high affinity and circulates at concentrations comparable to aldosterone. Severe deoxycorticosterone excess as is seen in 17α- and 11β-hydroxylase deficiencies causes hypertension, and moderate deoxycorticosterone overproduction in late pregnancy is associated with hypertension.
In addition, gene function enrichment analysis is carried out by using the g:Profiler, and the significant genes associated with hypertension are exhibited in Supplemenetary Table S6. For example, gene EIF2AK1 has negative regulation of hemoglobin biosynthetic process and negative regulation of translational initiation by iron. Atsma et al.40 showed that hemoglobin level is positively associated with both systolic and diastolic blood pressures. Gene AKR1C3 has negative regulation of isoprenoid metabolic process. Balakumar et al.41 indicated that the inhibition of synthesis of isoprenoids mediates the upregulation of endothelial nitric oxide synthase, a key enzyme involved in the regulation of cardiovascular function, by statins that are widely used in the treatment of dyslipidemia and associated cardiovascular abnormalities including hypertension.
Discussion
The family-based study plays an important role in genome-wide association studies. The members in the same family are homogeneous in their genetic background and thus there are more chances to detect susceptibility loci. The TDT-like methods detect genetic variants based on the difference between the number of minor alleles transmitted to the affected offspring from heterozygous parents and that not transmitted. Under Mendelian inheritance and no association between genetic variants and the disease, this difference would be close to 0. Because of the low frequency of rare variants, some family-based studies use collapsing/pooling method to enhance the signals and then to improve the power. However, there are several limitations on the existing approaches. First, a large proportion of variants in a genetic region may be noncausal/neutral, and the inclusion of these noises would definitely affect the detection power. Second, the causal variants may have opposite directions of association with disease, and collapsing would cancel out their collective effect, leading to low power. Third, the genetic region usually consists of both common and rare variants, and a threshold should be introduced to differentiate them.
Trio, an affected child and two parents, is a standard form of family data. In this paper, we use the multisite genotypes of trios to construct the test statistics. For a trio, the two nontransmitted alleles from parents are regarded as the genotype of a pseudocontrol. Hence, every affected child has a paired pseudocontrol. There would be no significant difference between the distribution of genetic variants of affected children and that of pseudocontrols if all genetic variants in a region have no association with diseases. We use Kullback–Leibler divergence30 to measure the difference between these two distributions, and the test statistics are therefore constructed to detect the functional genetic variants. Two test statistics KLTT and cKLTT are proposed to detect the associations of variants, rare or common, with common diseases. KLTT measures the difference between relative frequencies of genetic variants for the affected children and pseudocontrols; meanwhile, cKLTT measures the difference between frequencies of copy numbers of variants for the affected children and pseudocontrols directly. The proposed tests have some fulfilling features. First, these methods have no assumptions on the association mode, and thus are model free. Second, they are applicable to the genotype data, and there is no need to infer the phase by using some software. Third, the proposed methods could handle both common and rare variants simultaneously, and thus it is not necessary to set a threshold to distinguish them. Moreover, they measure the difference between distributions of variants for the affected children and pseudocontrols that would have the deserved power when there are genetic variants with opposite association directions.
We design extensive simulations to evaluate the performance of KLTT and cKLTT, and to compare them with the existing methods.20, 25, 29 The results of simulations show that KLTT and cKLTT are almost the same and the most powerful in situations of no or moderate LD when the candidate genetic region consists of both rare and common variants. When involving only rare variants, TSSU in Preston and Dudbridge25 is the best in some scenarios. It is desirable that our proposed methods are the second most powerful and the difference between the first and second highest powers decreases with the increase of LD level. Among KLTT and cKLTT, the performance of KLTT is superior to cKLTT when both rare and common variants exist (see Figures 1 and 2); cKLTT is more powerful than KLTT when only rare variants exist and the causal variants have opposite association directions. In addition, the LD level could affect the powers of all testing methods. The strong LD between genetic variants with opposite effect directions could reduce the powers, whereas the strong LD between genetic variants with the same directions could increase the powers. Finally, we apply the proposed methods to analyze the FHS data. Several significant genes are detected, and most of them have been shown in association with hypertension by other researches, such as genes SORBS1, EIF2AK1, ACSM3 and AKR1C3, demonstrating the usefulness of our methods.
Notice that our current test statistics are applicable to the standard trios. The extension to other kinds of family data is warranted. For example, sibling pair data, parents with multiple affected children and even a general pedigree. For the affected and unaffected sibling pair data, although we could directly utilize KLTT and cKLTT to measure the difference therein, the pedigree structure information is valuable and should be taken into account in the construction of test statistic. Finally, although the permutation procedure is computationally extensive, it is flexible in accommodating complicated LD structure among multiple variants. The recombinations among them, if existing, should be addressed in future study.
References
Hirschhorn, J. N. & Daly, M. J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 6, 95–108 (2005).
Eichler, E. E., Flint, J., Gibson, G., Kong, A., Leal, S. M., Moore, J. H. et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 11, 446–450 (2010).
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Bodmer, W. & Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 40, 695–701 (2008).
Schork, N. J., Murray, S. S., Frazer, K. A. & Topol, E. J. Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 19, 212–219 (2009).
Ahituv, N., Kavaslar, N., Schackwitz, W., Ustaszewska, A., Martin, J., Hébert, S. et al. Medical sequencing at the extremes of human body mass. Am. J. Hum. Genet. 80, 779–791 (2007).
Ji, W., Foo, J. N., O'Roak, B. J., Zhao, H., Larson, M. G., Simon, D. B. et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 40, 592–599 (2008).
Iyengar, S. K. & Elston, R. C. The genetic basis of complex traits: rare variants or “common gene, common disease”. Methods Mol. Biol. 376, 71–84 (2006).
Chen, G., Yuan, A., Zhou, Y., Bentley, A. R., Zhou, J., Chen, W. et al. Simultaneous analysis of common and rare variants in complex traits: application to SNPs (SCARVAsnp). Bioinform. Biol. Insights 6, 177–185 (2012).
Lee, S., Wu, M. C. & Lin, X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics 13, 762–775 (2012).
Amos, C. I. Successful design and conduct of genome-wide association studies Hum. Mol. Genet. 16, 220–225 (2007).
Benyamin, B., Visscher, P. M. & McRae, A. F. Family-based genome-wide association studies. Pharmacogenomics 10, 181–190 (2009).
Laird, N. M. & Lange, C. Family-based designs in the age of large-scale gene-association studies. Nat. Rev. Genet. 7, 385–394 (2006).
Wu, M. C., Lee, S., Cai, T., Li, Y., Boehnke, M. & Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93 (2011).
Pan, W., Kim, J., Zhang, Y., Shen, X. & Wei, P. A powerful and adaptive association test for rare variants. Genetics 197, 1081–1095 (2014).
Spielman, R. S., McGinnis, R. E. & Ewens, W. J. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet. 52, 506–516 (1993).
Rabinowitz, D. & Laird, N. A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum. Hered. 50, 211–223 (2000).
De, G., Yip, W.-K., Ionita-Laza, I. & Laird, N. Rare variant analysis for family-based design. PLoS ONE 8, e48495 (2013).
Ionita-Laza, I., Lee, S., Makarov, V., Buxbaum, J. D. & Lin, X. Family-based association tests for sequence data, and comparisons with population-based association tests. Eur. J. Hum. Genet. 21, 1158–1162 (2013).
He, Z., O'Roak, B. J., Smith, J. D., Wang, G., Hooker, S., Santos-Cortez, R. L. P. et al. Rare-variant extensions of the transmission disequilibrium test: application to autism exome sequence data. Am. J. Hum. Genet. 94, 33–46 (2014).
Li, B. & Leal, S. M. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 83, 311–321 (2008).
Auer, P. L., Wang, G. & Leal, S. M. Testing for rare variant associations in the presence of missing data. Genet. Epidemiol. 37, 529–538 (2013).
Madsen, B. E. & Browning, S. R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 5, e1000384 (2009).
Pan, W. Asymptotic tests of association with multiple SNPs in linkage disequilibrium. Genet. Epidemiol. 33, 497–507 (2009).
Preston, M. D. & Dudbridge, F. Utilising family-based designs for detecting rare variant disease associations. Ann. Hum. Genet. 78, 129–140 (2014).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Zhu, Y. & Xiong, M. Family-based association studies for next-generation sequencing. Am. J. Hum. Genet. 90, 1028–1045 (2012).
Sha, Q. & Zhang, S. A novel test for testing the optimally weighted combination of rare and common variants based on data of parents and affected children. Genet. Epidemiol. 38, 135–143 (2014).
Choi, S., Lee, S., Nöthen, M. M., Lange, C., Park, T. & Won, S. FARVAT: a family-based rare variant association test. Bioinformatics 30, 3197–3205 (2014).
Kullback, S. & Leibler, R. A. On information and sufficiency. Ann. Math. Statist. 22, 79–86 (1951).
Turkmen, A. S., Yan, Z., Hu, Y.-Q. & Lin, S. Kullback-Leibler distance methods for detecting disease association with rare variants from sequencing data. Ann. Hum. Genet. 79, 199–208 (2015).
Davies, R. B. The distribution of a linear combination of chi square random variables. J. Roy. Stat. Soc. C App. 29, 323–333 (1980).
Mailman, M. D., Feolo, M., Jin, Y., Kimura, M., Tryka, K., Bagoutdinov, R. et al. The NCBI dbgap database of genotypes and phenotypes. Nat. Genet. 39, 1181–1186 (2007).
Hsiung, C., Chuang, L.-M. & Hsiao, C.-F. Human SORBS1 genetic variations contribute to insulin resistance, obesity, type 2 diabetes, and hypertension, (13 August 2003) US Patent App. 10/639,491.
Lafferty, A. R., Torpy, D. J., Stowasser, M., Taymans, S. E., Lin, J. P., Huggard, P. et al. A novel genetic locus for low renin hypertension: familial hyperaldosteronism type II maps to chromosome 7 (7p22). J. Med. Genet. 37, 831–835 (2000).
So, A., Duffy, D. L., Gordon, R. D., Jeske, Y. W., Lin-Su, K., New, M. I. et al. Familial hyperaldosteronism type II is linked to the chromosome 7p22 region but also shows predicted heterogeneity. J. Hum. Hypertens. 23, 1477–1484 (2005).
Boomgaarden, I., Vock, C., Klapper, M. & D⊙ring, F. Comparative analyses of disease risk genes belonging to the acyl-CoA synthetase medium-chain (ACSM) family in human liver and cell lines. Biochem. Genet. 47, 739–748 (2009).
Naoharu, I., Katsuya, T., Toshifumi, M., Jitsuo, H., Toshio, O., Koichi, K. et al. Association between SAH, an acyl-CoA synthetase gene, and hypertriglyceridemia, obesity, and hypertension. Circulation 105, 41–47 (2002).
Sharma, K. K., Lindqvist, A., Zhou, X. J., Auchus, R. J., Penning, T. M. & Andersson, S. Deoxycorticosterone inactivation by AKR1C3 in human mineralocorticoid target tissues. Mol. Cell. Endocrinol. 248, 79–86 (2006).
Atsma, F., Veldhuizen, I., de Kort, W., van Kraaij, M., Pasker-de Jong, P. & Deinum, J. Hemoglobin level is positively associated with blood pressure in a large cohort of healthy individuals. Hypertension 60, 936–941 (2012).
Balakumar, P., Kathuria, S., Taneja, G., Kalra, S. & Mahadevan, N. Is targeting eNOS a key mechanistic insight of cardiovascular defensive potentials of statins? J. Mol. Cell Cardiol. 52, 83–92 (2012).
Acknowledgements
We thank two anonymous reviewers for their constructive comments and suggestions that improve the presentation of the manuscript greatly. We thank the FHS participants and acknowledge support from N01-HC25195. This work was supported in part by National Natural Science Foundation of China (11571082, 11171075), National Basic Research Program of China (2012CB316505) and the Scientific Research Foundation of Fudan University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, C., Sun, L., Zheng, H. et al. Detecting multiple variants associated with disease based on sequencing data of case–parent trios. J Hum Genet 61, 851–860 (2016). https://doi.org/10.1038/jhg.2016.63
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.63

{kind=link}