
Abstract
Exon skipping therapy has recently received attention for its ability to convert the phenotype of lethal Duchenne muscular dystrophy (DMD) to a more benign form, Becker muscular dystrophy (BMD), by correcting the open reading frame. This therapy has mainly focused on a hot-spot (exons 45–55) mutation in the DMD gene. Exon skipping of an entire stretch of exons 45–55 is an approach applicable to 46.9% of DMD patients. However, the resulting phenotype is not yet fully understood. Here we examined the clinical profiles of 24 patients with BMD resulting from deletions starting at exon 45. The Δ45–55 group ranged in age from 2 to 87 years; no mortality was observed, and one patient was ambulatory at 79 years of age. The age at which patients became wheelchair-bound in the Δ45–48 group (18–88 years old) was approximately 50 years. Cardiomyopathy was well controlled by pharmaceuticals in both deletion groups. In contrast, the Δ45–47 and Δ45–49 groups exhibited more severe phenotypes than those with other mutations: the age at which patients in the Δ45–49 group became wheelchair-bound was around 30–40 years. Our study shows that clinical severity differs between each hot-spot deletion.
Similar content being viewed by others

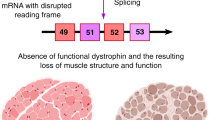

Introduction
Exon skipping therapy using antisense oligonucleotides has recently been proposed as a treatment for Duchenne muscular dystrophy (DMD), a lethal X-linked muscle disorder.1 DMD is caused by a mutation in the DMD gene that encodes the sarcolemmal protein, dystrophin. The mutation causes a disruption in the open reading frame (ORF), known as an out-of-frame mutation, and prevents dystrophin from being expressed.2 Conversely, sometimes the ORF is preserved despite the presence of a mutation (that is, in-frame mutation), resulting in a truncated but still functional dystrophin and that leads to the comparatively benign disorder, Becker muscular dystrophy (BMD).2 Accordingly, antisense oligonucleotides have been used to restore the DMD ORF by causing skipping of the mutated exon(s) during translation and converting the severe DMD phenotype into a milder form, similar to BMD.3
Dystrophin is a rod-shaped structure consisting of four domains: an N-terminal actin-binding domain, a central rod domain, a cysteine-rich domain, and a C-terminal domain.1 The central rod domain is composed of 24 spectrin-like repeats with four hinges. It includes over 76% of the structure’s protein and contains the great majority of the deletions that cause BMD.4, 5 In particular, a significant number of DMD-causative mutations occur between exons 45 and 55, making this a mutation hot spot.6 We previously described the cases of three unrelated patients with a Δ45–55 who showed very mild skeletal muscle involvement and could ambulate late in their lives.7 Similarly, two reports independently described patients with the same deletion mutation who exhibited a very mild or asymptomatic phenotype.8, 9 These observations suggest that skipping the entire 45–55 stretch of exons, using multiexon skipping, is a feasible treatment for DMD and severe BMD cases that are caused by mutations in this region.8, 10 Indeed, based on the Universal Mutation Database (UMD-DMD; http://www.umd.be/DMD/), this multiexon skipping could be applicable to 69.2% of DMD patients with large deletions (⩾1 exon) and 46.9% of all DMD patients with various mutations (Table 1). Furthermore, this approach has been reported to restore the dystrophin expression at the sarcolemma and lead to recovery of muscle pathology and function in a DMD mouse model.11
Whether or not restoring the ORF will also ameliorate symptoms in more benign BMD cases still needs to be clarified. Very recently, molecular homology modeling revealed that the proteins resulting from an in-frame deletion consist of a structure similar to normal dystrophin (hybrid-repeat) and an unrelated structure (fractional-repeat), and disease progression in BMD patients appears to be dependent on the structure and associated with a specific structure of dystrophin by deletion pattern.12 This report focused on deletion mutations Δ45–57, Δ45–48, Δ45–49 and Δ45–51;12 however, Δ45–55, which have attracted much attention, were not considered. We have already reported that truncated dystrophin with Δ45–55 is associated with a hybrid phenotype.11
Therefore, we also suggest that it is important to clarify the correlation between phenotype and genotype in BMD patients with in-frame mutations. In this study, we focused on the phenotype of patients with BMD, who had in-frame deletion starting at exon 45, to investigate the appropriate restoration of the reading frame by exon skipping therapy.
Materials and methods
A total of 43 patients with BMD were included in the study, who were receiving outpatient care from our hospital in Nagano Prefecture, Japan. The mutation analyses were performed by multiplex-ligation prove amplification, a combination of multiplex PCR and Southern blotting, or a combination of multiplex PCR and reverse transcription PCR (RT-PCR) using muscle biopsy specimens.5, 11, 12 Among them, the DMD mutation was identified in 35 patients, and we selected 24 patients harboring an in-frame deletion within the mutation hot spot, starting at exon 45. We examined their clinical profiles based on dystrophin conformation: hybrid type or fractional type, and performed a statistical analysis of the age at which patients became wheelchair-bound for each deletion mutation using the Kaplan–Meier method and log-rank test. Statistical significance was set at P<0.05. This study was approved by the institutional ethical committee of the Shinshu University School of Medicine, Japan (approval number 2270).
Results
Twenty-four patients with in-frame, hot-spot deletions were divided into groups based on the location of their deletions as follows: Δ45–57 (n=3), Δ45–48 (n=5), Δ45–49 (n=3), Δ45–51 (n=6), and Δ45–55 (n=7). We further divided the five deletion mutations into two groups, namely, hybrid type (Δ45–55, Δ45–48, and Δ45–51) and fractional type (Δ45–57 and Δ45–49) of dystrophin conformation.
The clinical profiles of hybrid-type mutations are shown in Table 2. In patients having Δ45–55, the age range was 2–87 years and no mortality occurred. We have previously reported three patients (Nos. 5–7) with this deletion who exhibited a very mild phenotype,5 and we have now added four younger patients (Nos. 1–4) as well. In the younger patients, the cause for diagnosis was hyperCKemia, and calf hypertrophy without muscle atrophy and weakness was observed.
The ages of the five patients with Δ45–48 ranged widely from 18 to 88 years. The 88-year-old patient died as a result of gall-bladder cancer. Three of these patients became wheelchair-bound around the age of 50 years. Cardiomyopathy was well controlled using angiotensin-converting enzyme inhibitor and/or a β-blocker. Our six patients with Δ45–51 are all young (<18 years) and have not yet shown any phenotype aside from hyperCKemia; therefore, we are continuing to care for their conditions, including cardiomyopathy.
The clinical profiles of fractional-type mutations (Δ45–47 and Δ45–49) are shown in Table 3. We had a small number of cases of these mutations (n=3, each); however, the age at which patients became wheelchair-bound in the Δ45–49 group was comparatively younger (30–40) and their cases were more severe than those of the hybrid-type form of dystrophin. Additionally, no mortality occurred and cardiomyopathy was controllable by pharmaceutical agencies, such as angiotensin-converting enzyme inhibitor or β-blockers.
We next examined the age at which patients became wheelchair-bound in each deletion group using Kaplan–Meier methods (Figure 1). With the exception of the Δ45–51 and Δ45–47 groups, in which patients were young and had not become wheelchair-bound, we performed log-rank tests between each group as follows: Δ45–55 vs Δ45–58, Δ45–55 vs Δ45–49, and Δ45–48 vs Δ45–49. We found a statistically significant difference between the Δ45–55 and Δ45–49 groups (P<0.05) and the Δ45–48 and Δ45–49 groups (P<0.05).

Kaplan–Meier analysis of the age at which patients became wheelchair-bound in each group: Δ45–55 (n=7), Δ45–48 (n=5), Δ45–49 (n=3), Δ45–47 (n=3), and Δ45–51 (n=6). The open circles show the age at which patients became wheelchair-bound; crossbars show the age at the non-wheelchair boundary but censored for this study. The log-rank test analyzed potential differences in the cumulative percentage of ambulatory patients between the two groups. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
The clinical phenotype of our cases having Δ45–55 was consistently very mild and this might be associated with the fact that Δ45–55 forms a hybrid-type dystrophin.11 Similar to Δ45–55 cases, patients with Δ45–58 also presented a relatively mild phenotype. This observation is consistent with a previous report,10 and this deletion also created a functionally stable hybrid-type dystrophin.10 The Δ45–51 created a hybrid type of dystrophin, indicating that this deletion might also result in a mild phenotype.
In contrast, the clinical phenotypes having fractional-type mutations Δ45–47 and Δ45–49 were more severe than the hybrid types. Despite our small number of patients, our study is the first to detect a significant difference in the degree of walking disturbance between the hybrid-type mutation (Δ45–55 or Δ45–58) and fractional-type mutation (Δ45–49) in Japanese BMD patients. It is important to note that the patient’s phenotypes could also depend on their environmental backgrounds and degree of exercise.
Regarding the cause of diagnosis, 16 of the 24 patients were diagnosed after hyperCKemia was detected in the asymptomatic stage. In patients with DMD, clinical severity and course are roughly homogenous because of a lack of functional dystrophin, and genetic counseling at the time of diagnosis has little effect. In contrast, the clinical severity and course of patients with BMD are heterogeneous and can depend on the type of mutation as well as other genetic or environmental factors. However, the specific details of these interactions are currently unknown. In the future, this information will be important for choosing the appropriate care and treatment courses for patients with BMD who are diagnosed at an early or asymptomatic stage. Furthermore, this knowledge will better inform clinical physicians when treating BMD.
Based on our results, in order to develop therapy focused on correcting the leading frame, the clinical phenotype that will result from the treatment should be considered. According to another report, Δ45–46 patients show typical DMD and the conformation of the resultant protein may result in protein instability or altered binding of critical partners.13 Thus, in DMD patients with Δ45, skipping exon 44 and multiexon skipping of exons 46 and 47 (or exons 46–48) are better potential therapies than skipping exon 46 alone; this report emphasizes that the development of exon skipping therapy must consider the genotype–phenotype correlation in dystrophinopathy.
Recently, not only exon skipping but also a CRISPR/Cas9 genome-editing strategy have received much attention as a way of correcting the leading frame. The CRISPR/Cas9 system can restore the DMD reading frame by targeting the mutational hot spot at exons 45–55 in DMD patient myoblasts.14 The CRISPR/Cas9 system can be used for a single large DMD deletion. Therefore, we would like to emphasize that larger cohort studies of BMD are necessary in order to provide a strategic rationale for the development of these therapies.
References
Hoffman, E. P., Brown, R. H. Jr. & Kunkel, L. M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 (1987).
Monaco, A. P., Bertelson, C. J., Liechti-Gallati, S., Moser, H. & Kunkel, L. M. An explanation for the phenotype differences between patients bearing partial deletions of the DMD locus. Genomics 2, 90–95 (1988).
Aartsma-Rus, A. & van Ommen, G. J. Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA 13, 1609–1624 (2007).
Tuffy-Giraud, S., Beroud, C., Leturcq, F., Yaou, R. B., Hamroun, D., Michel-Calemard, L. et al. Genotype-phnotype analysis in 2405 patients with a dystrophinopathy using UMD-DMD database: a model of nationwide knowledge-base. Hum. Mutat. 30, 934–945 (2009).
Nicolas, A., Lucchetti-Miganeh, C., Ben Yaou, R., Laplan, J. C., Chelly, J., Leturcq, F. et al. Assessment of the structural and functional impact on in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database. Orphanet J. Rare Dis. 7, 45 (2012).
Koenig, M., Beggs, A. H., Moyer, M., Scherpf, S., Heindrich, K., Bettecken, T. et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am. J. Hum. Genet. 45, 498–506 (1989).
Nakamura, A., Yoshida, K., Fukushima, K., Ueda, H., Urasawa, N., Koyama, J. et al. Follow-up of three patients with a large in-frame deletion of exons 45–55 in the Duchenne muscular dystrophy (DMD) gene. J. Clin. Neurosci. 15, 757–763 (2008).
Béroud, C., Tuffery-Giraud, S., Matsuo, M., Hamroun, D., Humbertclaude, V., Monnier, N. et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 28, 196–202 (2007).
Taglia, A., Petillo, R., D'Ambrosio, P., Picillo, E., Torella, A., Orsini, C. et al. Clinical features of patients with dystrophinopathy sharing the 45-55 exon deletion of DMD gene. Acta Myol. 34, 9–13 (2015).
Nakamura, A. & Takeda, S. Exon-skipping therapy for Duchenne muscular dystrophy. Neuropathol. 29, 494–501 (2009).
Aoki, Y., Yokota, T., Nagata, T., Nakamura, A., Tanihata, J., Saito, T. et al. Bodywide skipping of exons 45–55 in dystrophic mdx52 mice by systemic antisense delivery. Proc. Natl Acad. Sci. USA 34, 13763–13768 (2012).
Nicolas, A., Raguénès-Nicol, C., Ben Yaou, R., Ameziane-Le Hir, S., Chéron, A., Vié, V. et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet 24, 1267–1279 (2015).
Findlay, A. R., Wein, N., Kaminoh, Y., Taylor, L. E., Dunn, D. M., Mendell, J. R. et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann. Neurol. 77, 668–674 (2015).
Ousterout, D. G., Kabadi, A. M., Thakore, P. I., Majoros, W. H., Reddy, T. E. & Gersbach, C. A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 18, 6244 (2015).
Tasaki, N., Yoshida, K., Haruta, S. I., Kouno, H., Ichinose, H., Fujimoto, Y. et al. X-linked dilated cardiomyopathy with a large hot-spot deletion in the dystrophin gene. Intern. Med. 40, 1215–1221 (2001).
Yazaki, M., Yoshida, Y., Nakamura, A., Koyama, J., Nanba, T., Ohori, N. et al. Clinical characteristics of aged Becker muscular dystrophy wit onset after 30 years. Eur. Neurol. 42, 145–149 (1999).
Acknowledgements
This study was supported by an Intramural Research Grant (26-6) for Neurological and Psychiatric Disorders of the National Center of Neurology and Psychiatry (to AN). We thank Editage (www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Nakamura, A., Shiba, N., Miyazaki, D. et al. Comparison of the phenotypes of patients harboring in-frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J Hum Genet 62, 459–463 (2017). https://doi.org/10.1038/jhg.2016.152
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.152
This article is cited by
-
Cell Therapy Strategies on Duchenne Muscular Dystrophy: A Systematic Review of Clinical Applications
Stem Cell Reviews and Reports (2024)
-
Therapeutic approaches for Duchenne muscular dystrophy
Nature Reviews Drug Discovery (2023)
-
Duchenne muscular dystrophy
Nature Reviews Disease Primers (2021)
-
Genotype–phenotype correlation in Becker muscular dystrophy in Chinese patients
Journal of Human Genetics (2018)
