
Abstract
Whole-genome and -exome resequencing using next-generation sequencers is a powerful approach for identifying genomic variations that are associated with diseases. However, systematic strategies for prioritizing causative variants from many candidates to explain the disease phenotype are still far from being established, because the population-specific frequency spectrum of genetic variation has not been characterized. Here, we have collected exomic genetic variation from 1208 Japanese individuals through a collaborative effort, and aggregated the data into a prevailing catalog. In total, we identified 156 622 previously unreported variants. The allele frequencies for the majority (88.8%) were lower than 0.5% in allele frequency and predicted to be functionally deleterious. In addition, we have constructed a Japanese-specific major allele reference genome by which the number of unique mapping of the short reads in our data has increased 0.045% on average. Our results illustrate the importance of constructing an ethnicity-specific reference genome for identifying rare variants. All the collected data were centralized to a newly developed database to serve as useful resources for exploring pathogenic variations. Public access to the database is available at http://www.genome.med.kyoto-u.ac.jp/SnpDB/.
Similar content being viewed by others

Introduction
Next-generation sequencing technologies are revolutionizing the approach in identifying genetic variants that are associated with diseases. A current promising strategy focuses on rare variants that are shared among affected individuals but not found in public databases or appropriate normal control samples. This strategy is the most effective for rare Mendelian diseases, because it is unlikely that the rare pathogenic variants with large effect size have already been reported or deposited in public databases without linking to diseases. However, as these databases grow in size, the risk of eliminating genuine pathogenic variants that are segregating in the population will be augmented in the absence of a comprehensive knowledge of human genetic architectures including rare variants with their frequencies.
To expand our limited knowledge about genetic variation, several projects on whole-genome and -exome resequencing of a large number of individuals are now underway.1, 2, 3, 4, 5, 6 Such efforts include the international 1000 Genomes Project,2 which constructed a global reference of human genetic variation with a relatively small number of samples for each world-wide population. However, it is not sufficient as a source of population-specific rare and low frequency variants due to the limitation of coverage and sample number. The other large-scale sequencing efforts set their target population to Europeans or Africans. Among them, NHLBI GO Exome Sequencing Project has been collecting over 500 000 single-nucleotide variations on protein coding regions using the largest sample collections of >6500 European Americans and African Americans.5 According to these reports, the majority of the identified variants were previously unknown, population-specific and present at low allele frequencies, which suggests that a further exploratory survey of genetic variations in diverse populations will provide opportunities to better understand the role of genetic variations in the pathogenesis of human complex diseases as well as rare diseases.
In this study, we collected exomic sequencing data of 1208 Japanese individuals from five institutes and a data set of common variants determined by Illumina’s BeadArray technology from 3248 individuals of Japanese cohorts. Although several sequencing projects are going on for the East Asian population including Japanese for East Asian-specific genetic diversity information,4, 7, 8 our data sets currently provide the largest catalog of genetic diversity on the protein coding regions in the Japanese population. We centralized these data sets into a newly developed public database—human genetic variation database (HGVD). This database will serve as a useful resource sharing system, which will be required for research in genetic profiling in the future as well as for the development of genetic tests to screen for clinically relevant variants in personal genomes.
Materials and methods
Subjects
All subjects who participated in our exome sequencing projects were approved by the Institutional Review Board of each center (Kyoto University, National Research Institute for Child Health and Development, Tohoku University, University of Tokyo and Yokohama City University) in which donors gave written informed consent according to institutional and national guidelines, and anonymized to prevent the individual from being identified in the database. Each institute has ensured that all of the subjects have no clinical record associated with major diseases.
Exome sequencing and variation detection
Five data collection centers applied different combinations of exome capture and sequencing platforms on a subset of samples (Supplementary Table 1). Basic procedures are extraction of genomic DNAs from peripheral blood cells and enrichment of DNA fragments corresponding to exons using commercially available oligonucleotide libraries followed by applications to next-generation sequencers (HiSeq1000 (Illumina, San Diego, CA, USA), HiSeq2000 (Illumina) and SOLiD 5500XL (Thermo Fisher Scientific inc., Waltham, MA, USA)). The read lengths of the single- and paired-end libraries ranged from 75 to 101 bases. Each center processed the data using a variety of pipelines for variation call (Supplementary Table 2). The main functional steps of these pipelines were as follows: the sequences obtained from the individuals were aligned to the reference genome (NCBI Build 37/hg19) using the Burrows–Wheeler Aligner9 or Novoalign (http://www.novocraft.com/). Downstream processes included the removal of potential PCR duplicates, recalibration of base quality values, local realignment and variation call were analyzed using Samtools,10 Picard (http://broadinstitute.github.io/picard/) and GATK.11 The variations that were likely to be false -positive calls were filtered by optimization criteria drawn up at each center (Supplementary Table 2).
Identification of novel variations and functional prediction of genetic variations
Variations were categorized as novel if they were not registered in the dbSNP (Build 137),12 the 1000 Genomes Project (November 2010 data release),13 10 personal genomes (version 1.04)14 or the NHLBI GO Exome Sequencing Project (ESP6500SI).5 All identified variants were annotated using RefSeq15 and three types of deleteriousness scores were used to predict the effect of amino acid substitutions: (i) functional prediction score based on phylogenetic analysis and effect on protein structure with machine learning (PolyPhen-2),16 (ii) functional prediction score based on the degree of conservation of amino acid residues in sequence alignments (SIFT),17 and (iii) conservation score based on nucleotide sequence alignments and a model of neutral evolution (PhyloP).18 These scores were obtained from the dbNSFP database.19
Allele frequency spectrum
Under the assumption of neutral evolution at equilibrium, the expected number of sites at which the new nucleotide is present x times in the sample is given by 4Nμ/x, where N and μ are the effective population size and mutation rate, respectively.20 To compute the expected spectrum, 4Nμ is estimated from the observed segregating sites according to Watterson’s formula.21 In order to make comparisons to neutral without the effect of misidentification of ancestral states for these sites, the folded allele frequency spectra by minor allele count were projected. Only autosomal genes were included in calculating the allele frequency spectra.
Nucleotide diversity estimation
Pairwise nucleotide diversity (π) was calculated for all base pairs of RefSeq genes in autosomal regions. For this analysis, exomic sequences of 300 samples generated by Kyoto University were used. Only target base pairs with at least 10 × coverage were included. To evaluate departures from the expected patterns of neutral variation, we also calculated Tajima’s D, a statistic that compares nucleotide diversity estimated from the number of polymorphic sites observed in a given set of chromosomes against nucleotide diversity estimated from the allele frequency of the polymorphic sites for each gene.22
Fixation index calculation
We calculated unbiased estimates of fixation index (FST) as described previously.23 For the calculation, allele frequencies in African Americans and European Americans were downloaded from the Exome Sequencing Project website (http://evs.gs.washington.edu/EVS/, ESP6500SI).
Functional enrichment analysis for the genes having high FST and diversity
We used GeneTrail24 to test for enrichments of functional annotations for a set of 484 genes having high FST (⩾0.30) compared with African American versus European American (FST⩽0.30) and for a set of 46 genes having high-nucleotide diversity (π⩾0.005) values. The tests were performed against KEGG pathways,25 OMIM categories26 and gene ontology.27 We reported false discovery rates for each P-value using a hyper-geometric distribution.
Quality control and genetic analyzes for genotyping data set
Whole-genome genotyping was performed for a total of 3712 individuals, which is a subset of 9393 participants of The Nagahama Prospective Genome Cohort for the Comprehensive Human Bioscience (the Nagahama Study),28 using the Illumina HumanHap610 quad (Illumina), Human 2.5M (Illumina) and Human exome Beadarrays (Illumina). The 300 samples used for exome sequencing in Kyoto University were independently collected from the area of Nagahama city. However, they were given different anonymous ID, and due to an ethical reason, we are not able to link them with participants of the Nagahama Study. After excluding 162 samples of which genotyping call rates were smaller than 95%, kinship analysis and principal component analysis, were applied using PLINK (version 1.07)29 and SMARTPCA program of the EIGENSOFT package (version 2.0),30 respectively. There were 302 related individuals that were excluded from the further analysis, resulting in a data set of 3248 East Asian individuals, including 3 possible Chinese and 13 Korean individuals. (Supplementary Figure 1). A total of 1 794 196 single-nucleotide variations were included in the database after filtering out single-nucleotide variations with <99% genotyping success rates, with minor allele frequencies smaller than 0.01, or with Hardy Weinberg’s equilibrium P-values smaller than 1 × 10−7.
Evaluation of Japanese major allele reference sequence
In order to avoid biased evaluation of the Japanese major allele reference sequence, 192 Japanese exome sequencing data were randomly selected from in-house data sets and were used. The 100 base pair of paired-end reads from these samples were mapped onto the human genome reference sequence (Build 37/hg19) as well as the major allele reference sequences for African, European and Japanese populations using Burrows–Wheeler Aligner (version 0.6.1)9 with default parameters. Uniquely mapped reads are defined by the Burrows–Wheeler Aligner with which ‘XT:U’ tags are added in output files.
Results
Japanese genetic variation database
The HGVD is a web-accessible resource of genetic variations of the Japanese population. Currently, the database contains 287 588 single-nucleotide variations identified by whole-exome sequencing of 1208 individuals and 1 794 196 variants by genome scan of 3248 individuals with no record of major diseases. The HGVD is freely accessible via the internet at http://www.genome.med.kyoto-u.ac.jp/SnpDB. The database is developed with PostgreSQL (version 9.1.3) relational database, and the graphical user interface developed on JBrowse 1.4 (http://jbrowse.org/) is accessible on an Apache 2.2.3 server (https://httpd.apache.org/). The data in HGVD can be retrieved through the interface that allows users to search with a specific gene name or a dbSNP rsID and for a genomic region of interest. All of the HGVD data sets described in the following sections are downloadable as a series of text files that include allele frequency, genotype frequency and a Japanese major allele reference genome.
Exome sequencing
We sequenced 1208 healthy Japanese individuals. A total of 12.9 terabases of DNA sequence were generated and processed with a variety of analysis pipelines at each center (Supplementary Table 2). On average, 95.4% of the reads were mapped on the reference genome, which corresponded to 96.3% of the targeted bases covered with at least 10 × depths (Supplementary Table 1).
Allele frequency spectrum and functional impact of Japanese genetic variations
We identified 287 588 single nucleotide variants from the filtered data set of which 130 966 (45.5%) variants were found in the public database. Although the minor allele frequencies of the majority of the newly identified variants (139 096 or 88.8%) were smaller than 0.5%, the other 17 526 variants were found to be having minor allele frequency of greater than 0.5% (Figure 1a and Supplementary Figure 2). These observations indicate that not only the abundance of population-specific variation were rare, but also the common variations that are not registered in public databases are still found. The allele frequency spectrum of the Japanese population showed an excess of rare variations in comparison with the frequency spectrum predicted under the neutral equilibrium model (Figure 1b). The tendency was similar to European Americans rather than African Americans (Supplementary Figure S9 in Tennessen et al.5). Due to the abundance of rare variations, the mean score of Tajima’s D statistics22 was also negative (−0.95). These variation spectra in Japanese have not been clearly observed by the relatively small samples of exome sequencing analysis.31 On the other hand, a possible trace of recent rapid population expansion in demographics was suggested from the whole-genome sequencing analysis of 1070 Japanese individuals collected from a relatively small area.4

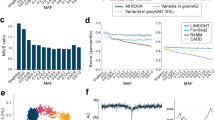
Frequency and functional spectrum of variations in the Japanese. (a) The proportion of newly identified non-synonymous, synonymous substitution and known variations in coding regions are indicated in red, green and gray bars, respectively. Known variations were defined as those that were previously reported in the public databases. (b) Allele frequency spectrum of variations in the Japanese. Allele frequency spectrum is shown with expected spectrum under a standard neutral model. (c) The proportions of identified non-synonymous (dark red) and synonymous (pink) substitutions are plotted against each bin of minor allele frequency in log scale. (d) Relationship between functional prediction scores and minor allele frequency. Fractions of functional damaging scores predicted from the three algorithms, i.e., PolyPhen-2, SIFT and PhyloP, are plotted for each bin of minor allele frequency in log scale.
To evaluate the functional impact of variations found in Japanese, we used four measures; categories of synonymous and non-synonymous (NS:S), PolyPhen-2,16 SIFT17 and PhyloP.18 In accordance with previous reports,5, 32 we observed an increased fraction of deleterious non-synonymous variations with lower minor allele frequencies (Figures 1c and d), suggesting that such variations arose recently enough to escape from purges of negative selection pressures.
Signatures of natural selection in the Japanese genome
The FST, a measure of genetic population differentiation,33 has proven to be a useful parameter for the ability to detect chromosomal loci that have been affected by natural selection.23 To identify candidate genes that may have been subjected to natural selection in Japanese, we calculated the unbiased estimates of FST using the allele frequency data from African American, European American and Japanese (FST-AEJ) and compared them with the FST from African American and European American (FST-AE). As shown in Figure 2, the majority of FST values were globally unchanged between two calculations (distributed along diagonal), because most of the variations in Japanese are likely to have similar frequencies to either African American or European American. Nevertheless, we noted that some fractions of the values were deviated from the diagonal with tendency toward higher in FST-AEJ rather than FST-AE, resulting from changes of allele frequency in Japanese population. Over representation analysis of the gene set having highly deviated FST values revealed an enrichment of well-known pigmentation genes MC1R and OCA2 (P=8.88 × 10−4). These genes are not only involved in melanin synthesis responsible for brown/lighter color of eyes and hair34 but also associated with oculocutaneous albinism (MIM 203200). High frequencies of derived alleles (rs1800414 and rs885479) of these regions were observed in Japanese (78.97 and 56.40%) compared with European Americans (4.82 and 0.03%) and African Americans (1.53 and 0.09%). The results support the signatures of recent positive selection, which were observed as significant extensions of haplotype homozygosity of these gene regions.35, 36 For the KEGG pathway,25 11 genes that are involved in extracellular matrix receptor interaction pathway were significantly enriched (P=4.83 × 10−4; Supplementary Table 3). Consistent with previous reports,23 these genes were distributed throughout the genome (Supplementary Figure 3 and Supplementary Table 4).

Increase of FST by allele frequency of Japanese population. (a) FST values for three populations (African American, European American and Japanese) and two populations (African American and European American) are plotted. Genes that have extremely higher values of FST from three population compared with those values from two populations are indicated in red dots in a and also shown with their gene symbols in b.
Mean nucleotide diversity (π) of Japanese was 0.042, which is higher than European American but lower than African American.5 As shown in Figure 3, genes related to immune function (HLA, CD52 and DEFB108B), olfactory receptors and ABO blood group, showed higher nucleotide diversity than other genes. The recompiled data allowed us to confirm the target of balancing selection to adapt a variety of pathogen and whose diversities are common characteristics across populations.5, 37

Genome-wide distribution of nucleotide diversity across genes in the Japanese. The value of π for each gene is shown as a vertical line. Genes that have high π values (>0.005) are shown by their gene symbol.
Construction of Japanese major-allele reference sequence
It has been shown that the ethnicity-specific major-allele reference sequence could improve genotyping accuracy for disease-associated variant loci.38 To apply this strategy for Japanese genomes, we substituted 816 991 positions of single nucleotide at the reference genome by the Japanese-specific major allele. By using 100-bases paired-end reads of independent exomic resequencing data, we were able to uniquely map 0.045% more reads to the Japanese-specific major allele reference sequence genome than to the NCBI reference sequence (Figure 4), due to the reduction in inconsistency of alignments (Supplementary Figure 4 and Supplementary Table 5).

Improved mapping rate of major-allele reference sequence of the Japanese. Short read data from exome sequence were mapped to original reference sequence (Build 37/hg19) and major-allele reference created using the resources from the NHLBI Exome Sequencing Project (ESP), the HapMap project, the 1000 Genomes project (1KG), and exome sequencing data of the current study. Numbers of individuals are 4300 for European American, 2200 for African American, 98 for Japanese and 1208 for Japanese reference, respectively. Mean percent increase of mapping rates are shown with s.d.
Discussion
We have developed a new database, HGVD, to provide the frequencies of genetic variations, which were determined by exome sequencing of 1208 healthy Japanese individuals. In addition to the above main component, the frequencies of common variants determined with genome scan of 3248 healthy Japanese are also accessible through the HGVD interface. For further genomics studies, their individual genotypes are available through the National Bioscience Database Center (http://humandbs.biosciencedbc.jp/en/, hum0012). Furthermore, we have also integrated the results of expression-QTL analysis, based on transcriptome data from individuals whose genotyping and exome sequencing data are available.39 This integrative data will help functional interpretations of genetic variations. Considering that the integrated information is not yet available from public databases for Japanese as compared with the African and European populations,5, 32, 40 the data set in HGVD will be informative not only for narrowing down mutations responsible for familial diseases, but also for explaining a part of missing heritability of complex diseases.41
The unique collection of high-resolution exonic variations in this study allows us to interpret the genetic diversity of the Japanese population by comparison with the data of European and African populations: low-allele frequency and high deleteriousness of the newly identified variations, excess of rare variations probably due to the recent rapid population growth in demographics, high-nucleotide diversity of immune related genes, and high frequency of derived allele in genes responsible for pigmentation and extracellular matrix receptor interaction. These signs of local adaptation can be regarded as an interesting guide for further evaluation of population-specific phenotype and disease prevalence study across populations.
Our catalog represented a snapshot based on currently available data generated from five institutes using non-uniform experimental procedures and analysis pipelines. The integration of these multiple data sets could be a disadvantage with respect to the uniformity of coverage or quality controls. Nevertheless, sequencing with more than a single platform and multiple libraries is vital to retain a high level of accuracy, as it creates a buffer against specific errors.42 Each institute deposited the data that passed their own quality criteria such as filtering based on GATK best practice (Supplementary Table S1). As these criteria are widely used and accepted in various sequencing projects,1, 2, 3, 6 it is unlikely that many false positives or negatives are included in our data sets. Although it was not possible to describe the accuracy of these deposited data sets with a single metric, the data are satisfactory to use for narrowing-down mutations responsible for rare Mendelian diseases.
Current surveys of genetic variation largely depend on the sequence of human genome reference constructed by the international project in 2003.43 Although the effort to improve the reference genome is being continued,44, 45 a considerable proportion of the genomic variation that have been recently identified in terms of numbers and sizes has been shown to be difficult to compile into a single reference genome without addressing incompatibilities. Therefore, an effort to reconstruct a more complete and ethnically applicable version of the human genome reference sequence with the format for integrating a variety of variation will be essential to bring about a new era for future human genome studies. This is also an eminent step forward for further improvement of the pathogenic variation identification as well as future systematic integration of genomic data into the practice of medicine and genetic diagnosis in the clinical field.
References
Genome of the Netherlands Consortium Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 46, 818–825 (2014).
1000 Genomes Project Consortium, Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Gudbjartsson, D. F., Helgason, H., Gudjonsson, S. A., Zink, F., Oddson, A., Gylfason, A. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015).
Nagasaki, M., Yasuda, J., Katsuoka, F., Nariai, N., Kojima, K., Kawai, Y. et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun. 6, 8018 (2015).
Tennessen, J. A., Bigham, A. W., O’Connor, T. D., Fu, W., Kenny, E. E., Gravel, S. et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69 (2012).
UK10K Consortium, Walter, K., Min, J. L., Huang, J., Crooks, L., Memari, Y. et al. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015).
Wong, L. P., Ong, R. T., Poh, W. T., Liu, X., Chen, P., Li, R. et al. Deep whole-genome sequencing of 100 southeast Asian Malays. Am. J. Hum. Genet. 92, 52–66 (2013).
Zhang, W., Meehan, J., Su, Z., Ng, H. W., Shu, M., Luo, H. et al. Whole genome sequencing of 35 individuals provides insights into the genetic architecture of Korean population. BMC Bioinformatics 15 (Suppl 11), S6 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001).
Durbin, R. M., Abecasis, G. R., Altshuler, D. L., Auton, A., Brooks, L. D., Gibbs, R. A. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Reese, M. G., Moore, B., Batchelor, C., Salas, F., Cunningham, F., Marth, G. T. et al. A standard variation file format for human genome sequences. Genome Biol. 11, R88 (2010).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65 (2007).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods. 7, 248–249 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Siepel, A., Pollard, K. & Haussler, D . New methods for detecting lineage-specific selection. Proceedings of the 10th international conference on research in computational molecular biology (RECOMB 2006), 3909, 190–205 (2006).
Liu, X., Jian, X. & Boerwinkle, E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 32, 894–899 (2011).
Fu, Y. X. Statistical properties of segregating sites. Theor. Popul. Biol. 48, 172–197 (1995).
Watterson, G. A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7, 256–276 (1975).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Akey, J. M., Zhang, G., Zhang, K., Jin, L. & Shriver, M. D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 12, 1805–1814 (2002).
Backes, C., Keller, A., Kuentzer, J., Kneissl, B., Comtesse, N., Elnakady, Y. A. et al. GeneTrail—advanced gene set enrichment analysis. Nucleic Acids Res. 35, W186–W192 (2007).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
McKusick, V. A. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 80, 588–604 (2007).
Harris, M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R. et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 (2004).
Tabara, Y., Takahashi, Y., Kawaguchi, T., Setoh, K., Terao, C., Yamada, R. et al. Association of serum-free fatty acid level with reduced reflection pressure wave magnitude and central blood pressure: the Nagahama study. Hypertension 64, 1212–1218 (2014).
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A. & Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Marth, G. T., Yu, F., Indap, A. R., Garimella, K., Gravel, S., Leong, W. F. et al. The functional spectrum of low-frequency coding variation. Genome Biol. 12, R84 (2011).
Nelson, M. R., Wegmann, D., Ehm, M. G., Kessner, D. St, Jean, P., Verzilli, C. et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 337, 100–104 (2012).
Weir, B. S. Population Substructure. Genetic Data Analysis II, Sinauer Associates, Sunderland, MA., (1996).
Beleza, S., Johnson, N. A., Candille, S. I., Absher, D. M., Coram, M. A., Lopes, J. et al. Genetic architecture of skin and eye color in an african-European admixed population. PLoS Genet. 9, e1003372 (2013).
Martinez-Cadenas, C., Lopez, S., Ribas, G., Flores, C., Garcia, O., Sevilla, A. et al. Simultaneous purifying selection on the ancestral MC1R allele and positive selection on the melanoma-risk allele V60L in south Europeans. Mol. Biol. Evol. 30, 2654–2665 (2013).
Shimanuki, M., Abe, Y., Tamiya, G., Ueki, M., Hozumi, Y. & Suzuki, T. Positive selection with diversity in oculocutaneous albinisms type 2 gene (OCA2) among Japanese. Pigment Cell Melanoma Res. 28, 233–235 (2015).
Seltsam, A., Hallensleben, M., Kollmann, A. & Blasczyk, R. The nature of diversity and diversification at the ABO locus. Blood 102, 3035–3042 (2003).
Dewey, F. E., Chen, R., Cordero, S. P., Ormond, K. E., Caleshu, C., Karczewski, K. J. et al. Phased whole-genome genetic risk in a family quartet using a major allele reference sequence. PLoS Genet. 7, e1002280 (2011).
Narahara, M., Higasa, K., Nakamura, S., Tabara, Y., Kawaguchi, T., Ishii, M. et al. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS ONE 9, e100924 (2014).
The GTEx Consortium The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Ratan, A., Miller, W., Guillory, J., Stinson, J., Seshagiri, S. & Schuster, S. C. Comparison of sequencing platforms for single nucleotide variant calls in a human sample. PLoS ONE 8, e55089 (2013).
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Genovese, G., Handsaker, R. E., Li, H., Altemose, N., Lindgren, A. M., Chambert, K. et al. Using population admixture to help complete maps of the human genome. Nat. Genet. 45, 406–414 (2013).
International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004).
Acknowledgements
This work was supported by the Practical Research Project for Rare/Intractable Diseases (201238002A) from the Ministry of Health, Labour and Welfare of Japan. We acknowledge the Nagahama City Office and the nonprofit organization ‘Zeroji Club’ for their help in conducting the Nagahama Study. Genotyping data of Nagahama cohort were produced through the support of a University Grant and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science & Technology in Japan and a research grant from the Takeda Science Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Higasa, K., Miyake, N., Yoshimura, J. et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet 61, 547–553 (2016). https://doi.org/10.1038/jhg.2016.12
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.12
This article is cited by
-
Pediatric hypertrophic cardiomyopathy caused by a novel TNNI3 variant
Human Genome Variation (2024)
-
Application of the CDK9 inhibitor FIT-039 for the treatment of KSHV-associated malignancy
BMC Cancer (2023)
-
A missense variant at the RAC1-PAK1 binding site of RAC1 inactivates downstream signaling in VACTERL association
Scientific Reports (2023)
-
Variant spectrum of PIEZO1 and KCNN4 in Japanese patients with dehydrated hereditary stomatocytosis
Human Genome Variation (2023)
-
Difference in Clinical Phenotype, Mutation Position, and Structural Change of RNF213 Rare Variants Between Pediatric and Adult Japanese Patients with Moyamoya Disease
Translational Stroke Research (2023)
