
Abstract
AARS2 gene (NM_020745.3) mutations result in two different phenotypic diseases: infantile mitochondrial cardiomyopathy and late-onset leukoencephalopathy. The patient’s first symptoms appeared at the age of 18 years with behavioral changes and psychiatric problems. Some years later, extrapyramidal symptoms, cognitive impairment, nystagmus, dysarthria and pyramidal symptoms also developed. The brain magnetic resonance imaging (MRI) indicated extensive white matter abnormalities. The diagnosis of AARS2 gene mutations causing leukodystrophy was confirmed by genetic testing. Segregation analysis confirmed the compound heterozygous state of the patient. Histological examination of the biopsy did not prove specific pathological alterations. The clinical phenotype of our patient was compared with seven previously described patients suffering from leukoencephalopathy caused by AARS2 mutations. We have documented a new, nonsense AARS2 gene mutation (c.578T>G, p.Leu193*) and a known missense mutation (c.595C>T, p.Arg199Cys) associated with leukoencephalopathy in a male patient. Clinical features, imaging characteristics and genetic testing are presented, and histological data from an AARS2-related leukodystrophy patient are described for the first time.
Similar content being viewed by others

Introduction
Mitochondrial disorders are frequently caused by dysfunction of the oxidative phosphorylation system that results in energy deficit in the cells. This group of diseases is genetically heterogeneous as some of the mitochondrial proteins are encoded by the mitochondrial DNA, whereas the rest of them are translated from messenger RNAs transcribed from the nuclear genome. The mitochondrial DNA contains 22 mitochondrial transfer RNA (tRNA) and 2 mitochondrial ribosomal RNA encoding genes, in addition to 13 protein encoding genes that are subunits in the mitochondrial respiratory chain. Several proteins involved in mitochondrial translation, such as initiation, elongation and termination factors, tRNA-modifying enzymes and the mitochondrial aminoacyl-tRNA synthetases (ARS2) are encoded by nuclear genes.1
Two groups of aminoacyl-tRNA synthetases can be distinguished based on their mitochondrial or cytoplasmic localization. These enzymes are encoded by distinct nuclear genes, with the exception of GARS and KARS, because these genes encode the glycyl- and lysyl-tRNA syntethases in both positions. Aminoacyl-tRNA synthetases play a significant role in the initiation of translation by charging tRNAs with their appropriate amino acids. All of these proteins have a catalytic and an anticodon binding domain (see later aminoacylation domain), and some of them, including mitochondrial alanyl-tRNA synthetase, also have an editing domain that can deacylate mischarged amino acids.2, 3
Mitochondrial alanyl-tRNA synthetase protein is encoded by the nuclear gene AARS2. Mutations of this gene result in two different phenotypic disorders that have been previously described. Certain mutations particularly affect the heart, producing a fatal, early-onset cardiomyopathy,4, 5, 6, 7 whereas others are associated with onset leukoencephalopathy and ovarian failure in females without cardiac involvement.8, 9 Euro et al.7 stated that the cause of the distinct phenotypes is related to the activity of the aminoacylation, with cardiomyopathy resulting from a serious reduction in aminoacylation activity, and leukodystrophy resulting from only moderate reduction in activity.
To date, only seven patients have been identified with AARS2-associated leukoencephalopathy and only one of them is male.
Here we report a young Caucasian male patient with a leukodystrophy phenotype and a novel AARS2 gene mutation.
Case presentation
Patient history and clinical data
The patient is a 29-year-old male with an uneventful perinatal period. His grandparents, parents and his younger brother did not report any neurological or psychiatric problems. The parents noted that the patient’s behavioral changes began at the age of 18 years. At that time he exhibited an alteration of personality, mania and paranoid delusions. Therefore, he was admitted to a psychiatric unit, where he underwent antipsychotic treatment. Some years later, extrapyramidal symptoms appeared and they were attributed to the side effects of the antipsychotic drugs. Otherwise, his somatic and intellectual development was normal until age 24 years, when signs of cognitive impairment became obvious. Before this deterioration, the patient was an excellent student in secondary school and also in college. There was a rapid and progressive cognitive decline, and during the following 2 years he evolved acalculia, orientation problems and dysgraphia. Besides these abnormalities, the neurological examination revealed horizontal and vertical nystagmus, dysarthria, dysphagia and extrapyramidal symptoms, including rigor, hypo- and bradykinesia, pyramidal symptoms such as Babinski sign and brisk tendon reflexes and frontal release signs, including glabellar, palmomental and sucking reflex and palmar grasp. Ophthalmoscopy and cardiological investigation did not reveal any pathological signs. The patient is still alive and is currently bedridden because of the severe neurological deficits.
MRI abnormalities
The brain magnetic resonance imaging (MRI) represented extensive white matter abnormalities, predominantly present in the frontal and parietal lobe, with a relative sparing of the central region. The corpus callosum was affected as well, especially the frontal part. Pyramidal tract involvement is represented at the level of the internal capsule. In addition, the frontopontine fibers are specifically affected within the internal capsule and in the brainstem (Figures 1a–f).

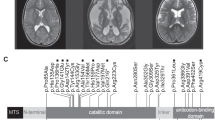
(a–f) Brain magnetic resonance imaging (MRI) of the patient, (g–i) AARS gene sequencing of the patient and his parents and (j) structure of the AARS2 gene. (a, b) T1-weighted, sagittal scans show corpus callosum involvement mainly in the frontal part and extended white matter abnormalities in the frontoparietal region with relative sparing of the central area. (c, d) T2-weighted, axial scans demonstrate pronounced leukoencephalopathy. The arrow proves the lesion of pyramidal tract in level of internal capsule. (e, f) Fluid-attenuated inversion recovery (FLAIR)-weighted, axial scans: the arrows show the frontopontine tract involvement in the level of internal capsule and the cerebral peduncle. AARS2 gene sequence of the patient (g) showing both mutations (c.578T>G and c.595C>T). DNA sequence of the patient’s father (h) and mother (i) demonstrating only one mutation, confirming the compound heterozygosity of the subject. Genomic structure of AARS2 (j) with exons coding for the aminoacylation (green), editing (yellow) and C-terminal (black) domains. The blue arrows (upper) indicate the position of mutations causing infantile mitochondrial hypertrophic cardiomyopathy. The lower arrows indicate the position of mutations causing leukoencephalopathy. The mutations of our patient indicated red color, whereas the other mutations resulting in leukodystrophy signed black color. The nonsense mutation (p.Leu193*) was reported here for the first time, whereas the missense mutation (p.Arg199Cys) has been described previously.5, 6, 8 A full color version of this figure is available at the Journal of Human Genetics journal online.
Laboratory findings
Numerous examinations were performed to determine the precise diagnosis of this early-adulthood-onset leukodystrophy. The laboratory findings, including a complete blood count, creatine kinase and serum lactate, were normal. The parameters of the cerebrospinal fluid, including leukocytes, protein content and IgG index, were within the normal range, and isoelectric focusing did not display any abnormalities. PCR tests of the herpes simplex virus types 1 and 2 from the cerebrospinal fluid and the HIV-1 test from blood were negative. The serum levels of very long chain fatty acids and phytanic acid were not elevated. The arylsulfatase-A and galactocerebrosidase activity from leukocytes showed intact function of these enzymes. The blood mass spectrometry for amino acids, acylcarnitine and succinylacetone did not point to metabolic deviations. Filipin staining in cultured fibroblasts did not confirm the diagnosis of Niemann–Pick disease type C.
Genetic tests
Genetic testing for the most common vanishing white matter disease causing gene EIF2B5 was also negative. Earlier genetic examinations did not verify CADASIL (Cerebral Autosomal-Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) disease. Based on the phenotype and the characteristic MRI features, targeted gene sequencing of AARS2 (NM_020745.3) was performed. The sequencing identified three variations: c.578T>G, c.595C>T and c.2188G>A (Figure 1g). The c.578T>G single-nucleotide polymorphism can be considered a pathogenic variation as it causes a nonsense mutation (p.Leu193*) in exon 3 and is likely to result in messenger RNA degradation by nonsense-mediated decay. This mutation was not described earlier and it could not be found in the ExAC database (The Exac Database; http://exac.broadinstitute.org/gene/ENSG00000124608). The c.595C>T variation causes a missense mutation (p.Arg199Cys) in exon 4 of the gene and its presence was also detected in four unrelated patients with similar clinical presentation.8 The frequency of this allele is 14/72 800 in the European population (The Exac Database; http://exac.broadinstitute.org/variant/6-44278885-G-A). The c.2188G>A single-nucleotide polymorphism also results in a missense mutation (p.Val730Met), but it is considered a polymorphism based on its high allele frequency in control subjects (∼4% in the European population; The Exac Database; http://exac.broadinstitute.org/variant/6-44270870-C-T). Segregation analysis indicated that the pathogenic mutations are not in the same allele of the AARS2 gene, as the patient’s father carries only the c.578T>G nonsense mutation, whereas his mother bears only the c.595C>T mutation. Consequently, the patient has a compound heterozygous mutation resulting in a late-onset leukodystrophy (Figures 1h and i). Figure 1j shows the aminoacylation, the editing and the C-terminal domains of the AARS2 gene with our patient’s and the earlier described mutations.
Histology
Before the identification of the genetic abnormality, a biopsy sampling from the frontal lobe, including cortex and white matter, was performed by another department. There was a lack of necrotic damage, inflammatory infiltration, neuronal changes or vascular lesions in the hematoxylin–eosin staining. In the Klüver–Barrera myelin staining, together with the immunostaining for myelin basic protein and neurofilament, we did not observe selective demyelination or axonal changes. Furthermore, there was a lack of amyloid precursor protein immunoreactive axonal bulbs. Immunostaining for CD3 (T cells), CD8 (cytotoxic T cells) and CD20 (B cells) was completely negative. There was an absence of macrophage activity (CD68) and only very minor microglial reaction was noted (HLA-DR). Screening for pathological protein inclusion using immunostaining for ubiquitin and p62 did not reveal unequivocal pathological deposits (Figure 2). In summary, this biopsy did not indicate pathological alterations, in particular not selective demyelination or axonal/myelin pathology.

Representative histopathological images of the biopsy sample. (a) Hematoxylin and eosin (HE) staining of the cortex reveals preserved neurons. HE (b), Klüver–Barrera (c), immunostaining for myelin basic protein (d), HLA-DR (microglia marker) (e) and SMI-31 (neurofilament) (f) of the white matter does not show disease-specific alterations, inflammation or demyelination. A full color version of this figure is available at the Journal of Human Genetics journal online.
Ethics approval and consent to participate
This study was approved by the ethical committee of the Faculty of Medicine, University of Szeged. Written informed consent was obtained from the patient for publication of this case report for educational purposes.
Discussion
In this paper we describe a patient with an early-adulthood-onset leukodystrophy caused by a compound heterozygous mutation of the AARS2 gene. One of these pathogenic mutations (c.578T>G) has not been previously published, whereas the other (c.595C>T) has been detected in four unrelated patients with leukodystrophy.8 To date, AARS2 mutations have been reported in 11 subjects who have infantile mitochondrial hypertrophic cardiomyopathy with early fatal outcomes and 7 patients with late-onset leukodystrophy.4, 5, 6, 7, 8, 9 Our patient’s phenotype is consistent with the predictions described by Euro et al.7 in 2015 that one nonsense mutation together with the milder p.Arg199Cys mutation cause the phenotype of leukodystrophy. Similar to the previous observation, our patient did not have any cardiac symptoms and his symptoms began after childhood and progressed rapidly. Behavioral changes were observed first and then neurological symptoms appeared, including progressive cognitive decline, frontal lobe dysfunction, cerebellar and extrapyramidal signs. The brain MRI abnormalities supported these symptoms, with extensive white matter lesions of frontal and parietal predominance, whereas corpus callosum, brainstem, pyramidal tract and frontopontine fiber involvement were observed. The clinical and radiological findings of our patient are similar to the subjects whose AARS2 gene mutation caused leukodystrophy (Table 1). As the biopsy sample included a region that was not severely affected, histopathological analysis did not reveal any disease-specific alteration.
In summary, ovarian failure and late-onset leukoencephalopathy raise the suspicion of the diagnosis of AARS2-associated leukodystrophy and vanishing white matter disease. The characteristic MRI abnormalities, the described neurological symptoms and the progressive course of the disease could help in differentiation. This novel form of leukodystrophy is extremely rare, with our patient being the eighth patient in the literature and only the second male patient. In this report we have identified a new, not previously published disease-causing mutation and the very first histological data of this disease.
References
Watanabe, K. Unique features of animal mitochondrial translation systems. The non-universal genetic code, unusual features of the translational apparatus and their relevance to human mitochondrial diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 86, 11–39 (2010).
Konovalova, S. & Tyynismaa, T. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol. Genet. Metab. 108, 206–211 (2013).
Diodato, D., Ghezzi, D. & Tiranti, V. The mitochondrial aminoacyl tRNA synthetases: genes and syndromes. Int. J. Cell Biol. 2014, 787956 (2014).
Götz, A., Tyynismaa, H., Euro, L., Ellonen, P., Hyötyläinen, T., Ojala, T. et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 88, 635–642 (2011).
Calvo, S. E., Compton, A. G., Hershman, S. G., Lim, S. C., Lieber, D. S., Tucker, E. J. et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 4, 118ra10 (2012).
Taylor, R. W., Pyle, A., Griffin, H., Blakely, E. L., Duff, J., He, L. et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312, 68–77 (2014).
Euro, L., Konovalova, S., Asin-Cayuela, J., Tulinis, M., Griffin, H., Horvath, R. et al. Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation. Front. Genet. 6, 21 (2015).
Dallabona, C., Diodato, D., Kevelam, S. H., Haack, T. B., Wong, L. J., Salomons, G. S. et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 82, 2063–2071 (2014).
Hamatani, M., Jingami, N., Tsurusaki, Y., Shimada, S., Shimojima, K., Asada-Utsugi, M. et al. The first Japanese case of leukodystrophy with ovarian failure arising from novel compound heterozygous AARS2 mutations. J. Hum. Genet. (e-pub ahead of print 2 June 2016; doi:10.1038/jhg.2016.64).
Acknowledgements
We thank Professor Marjo S van der Knaap, VU University Medical Center, Amsterdam, The Netherlands, for her help. This work was supported by Hungarian Brain Research Program KTIA Grant No. 13 NAP-A-II/17, KTIA Grant No. 13 NAP-A-II/18 and NKFI Grant K-112294.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Szpisjak, L., Zsindely, N., Engelhardt, J. et al. Novel AARS2 gene mutation producing leukodystrophy: a case report. J Hum Genet 62, 329–333 (2017). https://doi.org/10.1038/jhg.2016.126
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.126
This article is cited by
-
AARS2-Related Leukodystrophy: a Case Report and Literature Review
The Cerebellum (2022)
-
Whole-exome sequencing reveals new potential genes and variants in patients with premature ovarian insufficiency
Journal of Assisted Reproduction and Genetics (2022)
-
Mitochondrial aminoacyl-tRNA synthetase disorders: an emerging group of developmental disorders of myelination
Journal of Neurodevelopmental Disorders (2019)
-
A novel compound heterozygous mutation in AARS2 gene (c.965 G > A, p.R322H; c.334 G > C, p.G112R) identified in a Chinese patient with leukodystrophy involved in brain and spinal cord
Journal of Human Genetics (2019)
-
An adolescence-onset male leukoencephalopathy with remarkable cerebellar atrophy and novel compound heterozygous AARS2 gene mutations: a case report
Journal of Human Genetics (2018)
