
Abstract
Dysregulation of transcription is associated with the pathogenesis of cardiovascular diseases, including congenital heart diseases and heart failure. However, it remains unclear how transcription factors regulate transcription in the heart and which genes are associated with cardiovascular diseases in humans. Development of genome-wide analyses using next-generation sequencers provides powerful methods to determine how these transcription factors and chromatin regulators control gene expressions and to identify causative genes in cardiovascular diseases. These technologies have revealed that transcription during heart development is elaborately regulated by multiple cardiac transcription factors. In this review, we discuss the recent progress toward understanding the molecular mechanisms of how transcriptional dysregulation leads to cardiovascular diseases.
Similar content being viewed by others

Introduction
Regulation of gene expression by transcription factors and chromatin regulators is fundamental for determining cell type-specific properties. Transcription factors bind to target genomic regions to modulate the activity of enhancers and promoters.1, 2 Chromatin regulators are also involved in this regulation through the modification of chromatin status.3, 4, 5 Mutations in these factors are known to cause cardiovascular diseases, including congenital heart diseases (CHDs).6, 7, 8, 9, 10, 11, 12, 13, 14, 15
Transcription factors recognize specific DNA motifs16 and regulate gene expression by recruiting chromatin regulators.17, 18 In heart development, many cardiac-specific and non-specific transcription factors generate transcription factor networks to organize gene expression patterns.19, 20 Three transcription factors have been shown to be particularly important:8, 9, 10, 11 NK2 transcription factor-related locus 5 (Nkx2-5, also known as cardiac-specific factor) is specific to the heart and has a homeodomain as DNA-binding domain;9 T-box transcription factor, Tbx5, is involved in heart development as well as specification of limb identity;8, 21 and Gata4 is a zinc finger transcription factor that recognizes GATA motifs and has a critical role in the development of many organs, including the heart.10, 11 These factors are associated with each other and act as the core of the same transcription network.6, 7, 8, 12 Moreover, mutations in these genes have been found in patients with CHDs such as ventricular septal defect and atrial septal defect (ASD),6, 13, 14, 15 which suggests that cardiac transcription factors network serve a very important role.
The nucleosome is a basic unit composed of 146 bp of DNA and histone octomers to pack genomic DNA into a small nucleus. DNA is wrapped onto histone octomers consisting of two molecules of each histone (H2A, H2B, H3 and H4).22 Because genomic DNA is tightly packed in nucleosomes, regulation of this structure is critical for control of gene expression. Indeed, DNA methylation and histone modifications are used to change chromatin status, a process known as epigenetic regulation.23 Histone modifications are highly divergent, while most DNA modification is based on methylation.24, 25, 26 Histone modifications include methylation, acetylation, ubiqutination, citrullination and phosphorylation.25 Histones have a long tail region on the N-terminus. Most residues of histone tails are modified. Many histone modifiers recognize specific target residues of histone tails and control modification status on these residues. Mutations in genes coding histone modifiers were recently found to lead to CHDs.18, 27, 28, 29 Combination of histone modifications and DNA methylation may provide information to other transcription regulating factors through changing binding affinity between chromatin and these factors.
It is required to identify the genome-wide-binding sites of transcription regulating factors and chromatin status for elucidating the mechanisms of transcription regulation during heart development. Massively parallel DNA sequencing is a recently developed DNA sequencing technology that has revolutionized medical science.30, 31 This technology allows us to analyze the genome-wide-binding regions of transcription regulating factors. ‘Next generation sequencers’ have been developed by several companies including 454 Life Sciences (Branford, CT, USA), Illumina (San Diego, CA, USA), Life technologies (Grand Island, NY, USA), Pacific Biosciences (Menlo Park, CA, USA) and Helicos Biosciences (Cambridge, MA, USA). For example, 5500xl (Life Technologies), one of next-generation sequencers, can analyze 75 bp+35 bp DNA templates >600M reads. As the human genome size is 3G bp, next-generation sequencers can easily cover the whole human genome, thus this sequencer is suitable to use for genome-wide analysis. Indeed, many methods are developed using next-generation sequencers, such as the combination with chromatin immunoprecipitation and sequencing (ChIPseq) and whole-transcriptome shotgun sequencing (RNA sequencing; RNAseq).4, 5, 30, 31, 32 These technologies allow us to identify genomic mutation and to analyze the molecular mechanisms of chromatin regulation. Using massively parallel DNA sequencing, we can determine where transcription regulating factors bind and how they control gene expression via chromatin status modulation, as well as which mutations are associated with cardiovascular diseases.
This new technology has recently revealed the molecular mechanisms of transcription regulation by cardiac transcription factors and identified several genes related to cardiovascular diseases. In this review, we provide a review of recent studies that explore the molecular mechanisms of transcription regulation in heart development and cardiovascular diseases.
Regulation of chromatin during heart development
ChIPseq and RNAseq
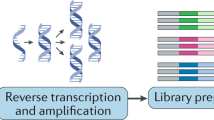
Identification of target genomic regions of transcription factors is key to understanding the functions of these factors. Prior to next-generation sequencing, it was extremely difficult to know the genome-wide-binding regions of these factors. ChIPseq permits us to identify these regions. We briefly illustrate the procedure for ChIPseq in Figure 1 (for additional detail, see these reviews by Metzker30Hawkins et al.31). Because next-generation sequencing generates several million reads in one run, bioinformatics is required to analyze the obtained DNA sequences.

Schematic illustration of ChIPseq and RNAseq. The procedure of ChIPseq. (1) Chromatin is digested by sonication or enzymes with/without crosslinking between proteins and DNA. (2) Complex of proteins of interest with their binding DNA is immunoprecipitated by the antibody against the protein of interest. (3) To make library of immunoprecipitated DNA, adapter DNA is ligated to the end of purified DNA, then these DNA fragments are amplified using the adapter DNA sequences. (4) DNA sequences in the library are analyzed by next-generation sequencing. Bioinformatic analysis begins with (1) mapping DNA sequences onto a reference genome, such as mm9 for the mouse genome and hg19 for the human genome. Several mapping tools are available from the next-generation sequencer company and researchers including Lifescope (Life technologies) and ELAND (Illumina), bowtie95 and BWA.96 (2) Mapped DNA sequences are then used for peak-calling to identify enriched genomic regions, but the results may vary based on tool and parameter settings, since peak-calling tools use various algorithms.97 (3) The obtained peaks are used for further analysis, such as comparing binding regions between samples, finding specific DNA motifs, and analyzing correlation with transcription status. The procedure of RNAseq. (1) Two different methods are available for mRNA collection: purification of polyA-tailed mRNA and depletion of rRNA. Because most cellular RNA is rRNA, depletion of rRNA is critical for enrichment of mRNA. Purification of polyA-tailed mRNA is easy to deplete rRNA, but non-polyadenylated mRNA, including histone mRNA, is lost.98 Alternatively, depletion of rRNA by sequence-specific probe is suitable to enrich mRNA, including non-polyadenylated mRNA, but more reads are required for analysis because sequence-specific probe cannot completely deplete rRNA. (2, 3) Purified mRNA is digested and used to construct a library, which is sequenced in the same manner as ChIPseq. Bioinformatic analysis begins with (1) the obtained reads are mapped onto a reference genome. Because splicing is occurred during maturation of mRNA, some reads are mapped in the junction between exons. Thus, it is required to assign mapping tools that could consider splicing, such as Tophat99, 100 and the programs from next-generation sequencer companies. (2, 3) Programs such as Cufflinks,99, 100 DESeq,101 bayseq102 or EdgeR,103 can be used to compare expression levels of mapped sequences between different samples. ChIPseq, chromatin immunoprecipitation and sequencing; RNAseq, RNA sequencing; rRNA, ribosomal RNA.
RNAseq is a method of RNA analysis. Many kinds of information about transcripts could be obtained depending on methods, such as gene expression level, transcription start site, transcription termination site, nascent transcripts and the length of polyA.33, 34, 35 mRNAseq is a method to analyze the mRNA population and most frequently used method in RNAseq. We briefly describe this procedure in Figure 1. The advantages of RNAseq are that it enables comparison of expression level between different genes and helps to obtain information on unknown genes' expression.
Dynamic changes of chromatin marks during cardiac differentiation from embryonic stem cells
Differentiation of stem cells into cardiac cells requires precise regulation of chromatin status to activate heart-related genes and repress genes for pluripotency. An experiment using embryonic heart is suitable to understand transition of chromatin status during cardiac differentiation. However, cardiac cells are a very small population in the early embryo, thus it may be difficult to collect sufficient cardiac cells to analyze chromatin status by ChIPseq. Alternatively, two studies show that embryonic stem cells (ESCs) can be used to study the transition of chromatin status during cardiac differentiation,36, 37 because cardiomyocytes are generated from ESCs.
Using mouse ESCs, Wamstad et al.36 defined the differentiation time to obtain the four cell types as ESCs, mesoderm, cardiac precursor and cardiomyocyte. They compared the difference of gene expression, histone modifications (active marks: H3K4me1, H3K4me3 and H3K27ac, and repressed mark, H3K27me3) and poised RNA polymerase II between these cell types (Table 1). In the activation of gene expression, including cardiac contracted genes that are substantially expressed in cardiomyocytes, H3K4me1 may be introduced into the locus for activation during transition of chromatin status. After this modification, H3K4me3 is introduced into a promoter, and RNA polymerase II is recruited. H3K4me1 is an enhancer marker.38, 39 These suggest that cardiac-specific enhancer is generated at the beginning of lineage commitments. Similarly to H3K4me1, another enhancer marker H3K27ac38, 39 is also introduced at the active enhancer regions. To identify which transcription factors are involved in the activation of cardiac enhancers, they searched DNA motifs at low-H3K27ac genomic regions in H3K4me1- and H3K27ac-marked active enhancers, because they thought that transcription factors bound to open-chromatin regions.36 OCT motif is found in ESCs and GATA, MEF and SRF motifs are found in differentiated cardiac cells. A combination of the transcription factors GATA and MEIS1A may be important to cardiogenesis-related genes activation.
A similar study using human ESCs also compared gene expression and histone modifications (H3K4me3, H3K27me3 and H3K36me3 that is one of the marker of transcribed genomic regions38).37 They defined the time course to obtain ESC, mesodermal progenitors, specified tripotential cardiovascular progenitors, committed cardiovascular cell and definitive cardiovascular cell.37 As expected, genes coding cardiac transcription factors are marked by H3K27me3 in ESCs. During cardiac differentiation, this H3K27me3 is decreased, while H3K4me3 and H3K36me3 are increased. On the other hand, structural proteins, including myosin heavy chain (MYH), are not marked by H3K27me3 in ESCs. This study also focused on MEIS, because its expression is increased along with differentiation. Indeed, knockdown of MEIS in zebrafish embryos perturbs cardiac morphogenesis.37 These two studies using mouse and human ESCs show the transition of chromatin status during differentiation of ESCs into cardiomyocytes and identifiy some transcription factors potentially involved in controlling this transition. However, how chromatin status is organized during cardiac differentiation is not yet fully understood. Recently, chromatin remodeling factor Brg1 is shown to be involved in the regulation of enhancers during cardiac differentiation.40 Although it is required to elucidate the transition of chromatin modifications in embryonic hearts, as cardiomyocytes differentiated from ESCs may not be similar to embryonic cardiomyocytes, these observations suggest an important role of chromatin modifications and remodeling to regulate gene expression during heart development.
Identification of cardiac enhancers
Identification of cardiac enhancers is one way to understand how cardiac gene expression pattern is constructed, as tissue-specific enhancers have a critical role to determine gene expression profiles. Enhancers are identified from a conserved sequence between different species in several tissues.41, 42, 43 Nevertheless, most of these conserved sequences do not work as cardiac enhancers,44 suggesting that the prediction of cardiac enhancers from the information of conserved DNA sequences is difficult at the present time. To explore cardiac enhancers, p300, a histone acetyltransferase, is used to identify enhancers, as p300 is known to bind to enhancers and to introduce acetylation to histones.44 p300-binding DNA was immnoprecipitated from 270 murine hearts at E11.5.44 ChIPseq of p300 identifies 3597 potential cardiac enhancers. In these regions, some p300-enriched regions are located near the heart-related genes. Approximately 84% of these regions are specific for the heart and 65% of these regions were conserved between placental mammals.44 Moreover, 81 out of 130 DNA sequences have the ability to activate expression in developing hearts in transgenic mice.44 This study shows that cardiac enhancers are unique in comparison with other tissue enhancers and p300 is useful to identify enhancers.
Visel and colleagues, who identified the mouse cardiac enhancers described above, also explored human cardiac enhancers.45 They found 10 059 p300-binding regions from human fetal heart at gestation week 16 and 4485 p300-binding regions from human adult heart. In these regions, 5047 p300-binding regions in fetal heart and 2233 regions in adult heart are located within 2.5 kb around transcription start sites. There were 1028 common p300-binding regions in fetal and adult hearts. Candidate human cardiac enhancers (43 out of 65) were evaluated in E11.5 transgenic mice. Only 21% of these putative cardiac enhancers are conserved between human and mouse, suggesting that the prediction of human cardiac enhancers from mouse data is difficult. In these studies, they used only p300 to identify cardiac enhancers. Information of chromatin status, including H3K4me1 and H3K27ac, could provide a finer map of cardiac enhancers in combination of p300-binding profile. Because H3K27ac-enriched chromatin regions are rapidly changed during heart development,46 suggesting that the activation of enhancers are elaborately controlled, it may be worth elucidating why heart has unique enhancers and how cardiac enhancers are regulated during development.
Transcription regulation in cardiovascular diseases
Gene regulation network in cardiomyocytes
Cardiac transcription factors including Nkx2-5, Tbx5 and Gata4 interact with each other and regulate heart development through regulation of gene expression, such as atrial natriuretic factor (ANF).6, 7 These transcription factors are well known to cause CHDs, including ASD and ventricular septal defect.6, 8, 10, 11, 13, 14, 15 However, how often these transcription factors coordinately bind to target genomic regions is unclear.
Recently, genome-wide-binding regions of several cardiac transcription factors have been identified by ChIP–chip and ChIPseq analyses. ChIP–chip is a method similar to ChIPseq, but analyses immunoprecipitated DNA by DNA-tiling arrays.31 Schlesinger et al.47 analyzed cardiac transcription factors, Gata4, Mef2a, Nkx2-5 and Srf, in cardiomyocyte cell line HL-1 by ChIP–chip. HL-1 is established from an atrial cardiomyocyte tumor of AT-1 mice that express SV40 large T antigen in atrial cardiomyocytes by the ANF promoter.48 They also explored the affected genes by knocking down of these transcription factors in HL-1 cells. Most of these transcription factor-binding sites are close to transcription start sites and 498 genes, including the genes coding structural proteins, are regulated by multiple transcription factors. Knockdown of these transcription factors affects the expression of genes coding for cardiac structure proteins that are bound by these transcription factors. Moreover, they found that Srf binding is correlated with histone H3 acetylation in actively transcribed genes.
He et al.49 also tried to identify genome-wide-binding regions of tagged cardiac transcription factors: Nkx2-5, Gata4, Tbx5, SRF and Mef2a, and p300 in HL-1 cells. They explored which transcription factors coordinately bound by DNA motif analysis in these cardiac transcription factor-binding regions. The motif of TEAD1 (TEA domain family member 1) is enriched around p300, Nkx2-5, Gata4 and Mef2a-binding regions. TEAD1 is a cardiac transcription factor that regulates muscle genes expression.50 Indeed, TEAD1 physically interacts with these factors.49 Analyzing ChIPseq data, 1715 multiple transcription factors (four and five transcription factors)-binding regions were found (Figure 2a). Although p300 does not bind to most of these regions, these regions have enhancer activity in transgenic mice (7 out of 13 loci). Consistent with this study, collective transcription factors occupancy was found to activate enhancer in Drosophila,1, 51 indicating conservation of this collective occupancy of transcription factors to activate enhancer across species.

Schematic illustration of the regulation of gene expression by cardiac transcription factors. (a) Cardiac transcription factors, including Nkx2-5, Gata4, Tbx5, SRF and Mef2a, collectively bind to enhancer. (b) Cardiac transcription factors form complex with chromatin regulators. (c) Stage- and environment-specific transcription factor (e.g., Gata4) binding to the target regions. Box shows the transcription factor-binding site. Nkx2-5, NK2 transcription factor-related locus 5; TF, transcription factor.
Cardiac transcription factors also regulate functions of the conduction system of heart through control of gene expression, including SCN10a, an ion channel protein.52 Tbx3, Gata4, Nkx2-5 and p300 are bound at the Scn10a gene locus in adult murine hearts.52 One of these binding regions may act as an enhancer of SCN10a. Indeed, a single-nucleotide polymorphism (SNP) at this region reduces Tbx3 binding and might affect cardiac function, suggesting that disease-related SNPs that do not change protein structure may affect the activity of enhancers by disrupting the transcription factor binding.
Relationships between transcription factors and chromatin regulators
Transcription factors require several chromatin regulators including histone modifiers and chromatin remodeling factors1, 28 (Figure 2b). Thus, it is critical to understand the molecular mechanisms of transcription factors to identify transcription factor-associated factors. In this section, we introduce several cofactors that regulate transcription with cardiac transcription factors.
Histone methyltransferases and demethyltransferases regulate the histone methylation pattern.25, 53 These enzymes have distinct target residues in histone proteins. As described above, the histone methylation pattern is highly correlated with chromatin status. Ash2L (Absent small homeotic 2) has the activity to catalyze H3K4me3 and interacts with the MLL (Mixed lineage leukemia) complex.54 Ash2L interacts with Tbx1, a causative gene for DiGeorge syndrome, and activates Tbx1-mediated transcription in a dose-dependent manner.55 Whsc1 (Wolf-Hirshhorn syndrome candidate 1, also known as NSD1 or MMSET) has the activity to catalyze H3K36me3 and functionally interacts with Nkx2-5.18 Whsc1 and Nkx2-5 complex regulate gene expression level, including Pdgfra, through the enzymatic activity of H3K36me3.18 Whsc1 also interacts with Runx2,56 suggesting that Whsc1 regulates transcription activity via transcription factors. Ezh2 has the activity to catalyze H3K27me3 and has critical role in heart development.29 Nkx2-5 associates with Jarid2 (also known as JMJ), which associates with the Ezh2 complex to repress gene expression.57, 58 These studies indicate that Nkx2-5 and its binding partners may have a key role in regulating chromatin structure and gene expression in cardiac cells.
Histone acetylation is highly correlated to active chromatin status and is regulated by histone acetyltransferases and deacetylases (HDAC).59 Some transcription factors, along with these enzymes, control histone acetylation. Tbx5 associates with p300 and p300/CBP-associated factor (PCAF), a histone acetyltransferase, through a WW-domain-containing transcriptional regulator TAZ (also known as WWTR1) and promotes ANF gene expression.60 Tip60, a histone acetyltransferase, is also associated with Tbx5.61 Acetylation of Nkx2-5 by p300 prevents the interaction between Nkx2-5 and p300.62 HDAC5 deacetylates from Nkx2-5, which promotes Ncx1 (sodium calcium exchanger) gene expression through the association between Nkx2-5 and p300. p300 is also involved in Gata4 complex.63, 64 Myocardial infarction induces acetylation in Gata4 by p300, which increases its DNA-binding ability. Acetylation has a critical role not only in transcription activation but also in control of interaction between Nkx2-5 and p300.
Chromatin remodeling factors Brg1 and Baf60c associate with Nkx2-5, Tbx5 and Gata4 to activate transcription of target genes.63, 65 Mutations in chromatin remodeling factors cause congenital diseases.27, 66 For example, mutations in CHD7, an ATP-dependent chromatin remodeling factor, causes CHARGE syndrome.67, 68 CHARGE syndrome is a congenital disease and the patients have multiple disorders, including CHDs, ocular coloboma of the eye, atresia of the choanae, retarded growth, genital hypoplasia and ear defects.67, 68 Recently, p53 was reported to be involved in the pathogenesis of CHARGE syndrome.69 CHD7 knockout slightly increases p53, and p53 heterozygousity partially rescues CHD7 knockout phenotypes. Mutations in p53, which lead to stabilization of p53 protein, are embryonic lethal and causes heart defects including DORV (Double outlet right ventricle), if wild-type p53 is present. Consistent with this report, another group has shown that increased p53 is critical for progression from hypertrophy to heart failure.70 These studies indicate the activation of p53 is involved in chromatin remodeling factor-related congenital diseases.
Transcription regulation in hypertrophy model mouse
Gata4 is involved in heart development and adult heart diseases.71 Adult heart disease induces expression of many genes in embryonic hearts,72 however, whether re-expressed cardiac transcription factors, including Gata4, bind to the same genomic regions as embryonic hearts is unclear. Thus, He et al.73 compared Gata4-binding regions among embryonic hearts, adult hearts, and pressure-overloaded hearts. Gata4-binding regions are strikingly different dependently on the status of heart (Figure 2c). Approximately 80% of embryonic Gata4-binding regions are specific in comparison with adult Gata4-binding regions. Pressure overload induces new Gata4-binding regions that are not bound in the embryonic heart, suggesting that Gata4 generates disease-related enhancers in pressure-overloaded hearts. Gata4 binding is found near actively transcribed genes, but there is no correlation between the strength of binding and gene expression. Although the mechanisms controlling Gata4-binding regions in pressure overload is not clear, other transcription factors such as NFAT may be associated with this mechanism, because NFAT motifs are found at the distal Gata4-binding regions in embryonic and pressure-overloaded hearts.
Recent developments with non-coding RNAs may explain the mechanisms in which Gata4-binding regions change across heart development. A non-coding RNA, Mhrt (Myosin heavy-chain-associated RNA transcripts), was recently reported to be involved in the progression of cardiomyopathy through inhibition of Brg1.74 Mhrt is reversely transcribed from the Myh7 gene locus and is increased along with the progress of heart development. Overexpression of Mhrt inhibits cardiomyopathy. Brg1 is critical for myocardial proliferation. Depletion of Brg1 inhibits cardiac hypertrophy that is induced by pressure overload.75 Mhrt interacts with Brg1 and prevents the function of Brg1. On the other hand, Brg1–HDAC–PARP complex represses Mhrt expression under pathological stress. These studies suggest that the change in chromatin structure has pivotal roles for the change in heart function.
The control of histone acetylation by p300 and HDACs is important for the progress of heart failure.76, 77, 78, 79 To understand the role of acetylated lysine reader, Anand et al.80 inhibited Brd4 by administration of selective bromodomain inhibitor, JQ1.81 Brd4, one of the BET family protein and highly expressed among BRD2, 3 and 4 in the heart, recognizes acetylated lysine through the Bromo domain.80 Administration of JQ1 inhibits the progress of heart failure. JQ1 prevents changes of global gene expression pattern, including GATA4 and NFκB expression, that are upregulated by pressure overload. JQ1 represses elongation of RNA polymerase II through inhibition of the function of p-TEFb, because BRD4 is associated with CDK9 that is a component of p-TEFb. These studies indicate that the pathway of histone acetylation is critical for the progress of heart failure. Inhibition of signaling of histone acetylation may be a good candidate for their treatment.
Genome-wide-associated studies for discovering the mutations in CHDs
Although mutations in some genes encoding transcription regulating factors are well known to cause CHDs,6 it was still unclear which genes are related to the pathogenesis of CHDs. Many cases are sporadically found, meaning that the identification of de novo mutations is important for understanding the mechanisms of pathogenesis of these diseases. Zaidi et al.82 identified several de novo mutations in H3K4 methylation-related genes by analyzing exome sequencing data of trios: 362 patients who have severe CHDs, including conotruncal defects, left ventricular obstruction and heterotaxy and their parents.82 They found mutations in H3K4 methylation-related genes, MLL2, WDR5, KDM5A, KDM5B and CHD7, in H2BK120 ubiquitination-related genes, RNF20, UBE2B, USP44, in SMAD2, and in other genes related to the regulation of transcription. Although the reason why mutations in the genes regulating H3K4 methylation, but not other histone modifications, are significantly enriched in CHDs is still unclear, it may be important to elucidate how the dysregulation of H3K4 methylation changes global gene expression pattern during heart development. Protein-altering mutations are found in around 10% CHD patients in their study. Mutations in non-coding regions, including enhancers, may be associated with the onset not only of CHDs but also of other diseases, including cancer.83, 84 Chromatin modifications without nucleotide changes may be associated with the disease onset.
Although exome sequencing reveals many de novo mutations in CHDs,82 analysis of SNPs related to CHDs by DNA microarray may be abstruse to find causative genes for the onset of CHDs. One group analyzed 5112 controls and 4225 CHDs, including ASD, ventricular septal defect and ASD/ventricular septal defect to avoid heterogeneity.85 Although they found two SNPs, these SNPs may not have a significant function in CHDs. Another group analyzed 1995 CHDs, including septal, obstructive and cyanotic defects and 5159 controls.86 They could not find significant SNPs from all CHDs data, but could find three SNPs in 4p16 that are causative regions for the onset of Wolf-Hirshhorn syndrome87 from ostium secundum ASD data. Although one of the SNPs is located at an EST in the intron of non-coding RNA that is expressed in human heart, it is unclear whether this SNP has a function. De novo mutations may be more related to the onset of CHDs than common SNPs.
Both exome sequencing and DNA microarray have different advantage and disadvantage points. Exome sequencing reveals de novo mutation in exon but not the mutations in introns and non-coding regions, while DNA microarray reveals genome-wide SNPs but not de novo mutations. To overcome these disadvantage points, Glessner et al.88 use both technologies to identify genome-wide copy number variations (CNVs) in 538 CHDs and 1301 controls. They found 63 de novo CNVs in 51 CHDs and identified both known CNVs and new CNV. Recurrent de novo CNVs in 15q11.2 as two deletions and two duplications include CYFIP1 (cytoplasmic FMR1-interacting protein 1).89 CYFIP1 is highly expressed during heart development, thus it may be related to heart development through the regulation of local protein synthesis and actin remodeling.90 They also found the mutations in specific genes, including ETS1 and CTBP2. ETS1 is a transcription factor that might be a causative gene for Jacobsen syndrome.91 CTBP2 acts as a transcriptional repressor and has an NAD+ binding domain.92 CTBP2 knockout mice exhibit CHDs.93 These results suggest that the frequency of de novo CNVs is increased in CHDs and that the dysregulation of transcription is implicated in the onset of CHDs.
Conclusion
Mutations in many genes coding transcription factors and chromatin regulators are related to the onset of cardiovascular diseases, including CHDs. The regulation of chromatin is implicated in cardiovascular diseases. However, it is still unclear whether genome-wide chromatin modifications, including histone modifications and DNA methylation, are changed in cardiovascular diseases and how dysregulation of chromatin regulation leads to these diseases is not fully elucidated. Moreover, it should be elucidated why specific heart regions, including the septum, are easily affected by transcriptional dysregulation. Because each heart region activates different enhancers during heart development,94 it is necessary to elucidate how gene expression is regulated in these regions by next-generation sequencing for understanding the mechanisms of the onset of CHDs.
References
Spitz, F. & Furlong, E. E. M. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626 (2012).
Chen, X., Xu, H., Yuan, P., Fang, F., Huss, M., Vega, V. B. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 (2008).
Ruthenburg, A. J., Li, H., Patel, D. J. & David Allis, C. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 8, 983–994 (2007).
Barski, A., Cuddapah, S., Cui, K., Roh, T., Schones, D., Wang, Z. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007).
Mikkelsen, T. S., Ku, M., Jaffe, D. B., Issac, B., Lieberman, E., Giannoukos, G. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 (2007).
Bruneau, B. G. The developmental genetics of congenital heart disease. Nature 451, 943–948 (2008).
Srivastava, D. Making or breaking the heart: from lineage determination to morphogenesis. Cell 126, 1037–1048 (2006).
Bruneau, B. G., Nemer, G., Schmitt, J. P., Charron, F., Robitaille, L., Caron, S. et al. A Murine Model of Holt-Oram Syndrome Defines Roles of the T-Box Transcription Factor Tbx5 in Cardiogenesis and Disease. Cell 106, 709–721 (2001).
Lyons, I., Parsons, L. M., Hartley, L., Li, R., Andrews, J. E., Robb, L. et al. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 9, 1654–1666 (1995).
Molkentin, J. D., Lin, Q., Duncan, S. A. & Olson, E. N. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 11, 1061–1072 (1997).
Kuo, C. T., Morrisey, E. E., Anandappa, R., Sigrist, K., Lu, M. M., Parmacek, M. S. et al. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 11, 1048–1060 (1997).
Garg, V., Kathiriya, I. S., Barnes, R., Schluterman, M. K., King, I. N., Butler, C. A. et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424, 443–447 (2003).
Basson1, C. T., Bachinsky, D. R., Lin, R. C., Levi, T., Elkins, J. A., Soults, J. et al. Mutations in human cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 15, 30–35 (1997).
Yi, Li, Q., Newbury-Ecob, R. A., Terrett, J. A., Wilson, D. I., Curtis, A. R. J., Ho, Yi, C. et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 15, 21–29 (1997).
Schott, J. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281, 108–111 (1998).
Pabo, C. O. & Sauer, R. T. Transcription factors: structural families and principles of DNA recognition. Annu. Rev. Biochem. 61, 1053–1095 (1992).
Voss, T. C. & Hager, G. L. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet. 15, 69–81 (2014).
Nimura, K., Ura, K., Shiratori, H., Ikawa, M., Okabe, M., Schwartz, R. J. et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 460, 287–291 (2009).
Olson, E. N. Gene regulatory networks in the evolution and development of the heart. Science 313, 1922–1927 (2006).
Buckingham, M., Meilhac, S. & Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 6, 826–837 (2005).
Rallis, C., Bruneau, B. G., Buono, J. D., Seidman, C. E., Seidman, J. G., Nissim, S. et al. Tbx5 is required for forelimb bud formation and continued outgrowth. Development 130, 2741–2751 (2003).
Luger, K., Mäder, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997).
Goldberg, A. D., Allis, C. D. & Bernstein, E. Epigenetics: a landscape takes shape. Cell 128, 635–638 (2007).
Plongthongkum, N., Diep, D. H. & Zhang, K. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat. Rev. Genet. 15, 647–661 (2014).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Holliday, R. & Pugh, J. E. DNA modification mechanisms and gene activity during development. Science 187, 226–232 (1975).
Bruneau, B. G. Chromatin remodeling in heart development. Curr. Opin. Genet. Dev. 20, 505–511 (2010).
Nimura, K., Ura, K. & Kaneda, Y. Histone methyltransferases: regulation of transcription and contribution to human disease. J. Mol. Med. 88, 1213–1220 (2010).
Delgado-Olguín, P., Huang, Y., Li, X., Christodoulou, D., Seidman, C. E., Seidman, J. G. et al. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat. Genet. 44, 343–347 (2012).
Metzker, M. L. Sequencing technologies—the next generation. Nat. Rev. Genet. 11, 31–46 (2010).
Hawkins, R. D., Hon, G. C. & Ren, B. Next-generation genomics: an integrative approach. Nat. Rev. Genet. 11, 476–486 (2010).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Ozsolak, F. & Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 12, 87–98 (2011).
Core, L. J., Waterfall, J. J. & Lis, J. T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848 (2008).
Chang, H., Lim, J., Ha, M. & Kim, V. N. TAIL-seq: genome-wide determination of poly(A) tail length and 3′ end modifications. Mol. Cell 53, 1044–1052 (2014).
Wamstad, J. A., Alexander, J. M., Truty, R. M., Shrikumar, A., Li, F., Eilertson, K. E. et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 151, 206–220 (2012).
Paige, S. L., Thomas, S., Stoick-Cooper, C. L., Wang, H., Maves, L., Sandstrom, R. et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 151, 221–232 (2012).
Zhou, V. W., Goren, A. & Bernstein, B. E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 12, 7–18 (2011).
Levine, M., Cattoglio, C. & Tjian, R. Looping back to leap forward: transcription enters a new era. Cell 157, 13–25 (2014).
Alexander, J. M., Hota, S. K., He, D., Thomas, S., Ho, L., Pennacchio, L. A. et al. Brg1 modulates enhancer activation in mesoderm lineage commitment. Development 142, 1418–1430 (2015).
Woolfe, A., Goodson, M., Goode, D. K., Snell, P., McEwen, G. K., Vavouri, T. et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 3, e7 (2004).
Prabhakar, S., Poulin, F., Shoukry, M., Afzal, V., Rubin, E. M., Couronne, O. et al. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 16, 855–863 (2006).
Pennacchio, L. A., Ahituv, N., Moses, A. M., Prabhakar, S., Nobrega, M. A., Shoukry, M. et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 (2006).
Blow, M. J., McCulley, D. J., Li, Z., Zhang, T., Akiyama, J. A., Holt, A. et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 42, 806–810 (2010).
May, D., Blow, M. J., Kaplan, T., McCulley, D. J., Jensen, B. C., Akiyama, J. A. et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 44, 89–93 (2012).
Nord, A. S., Blow, M. J., Attanasio, C., Akiyama, J. A., Holt, A., Hosseini, R. et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531 (2013).
Schlesinger, J., Schueler, M., Grunert, M., Fischer, J. J., Zhang, Q., Krueger, T. et al. The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet. 7, e1001313 (2011).
Claycomb, W. C., Lanson, N. A., Stallworth, B. S., Egeland, D. B., Delcarpio, J. B., Bahinski, A. et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl Acad. Sci. USA 95, 2979–2984 (1998).
He, A., Kong, S. W., Ma, Q. & Pu, W. T. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl Acad. Sci. USA 108, 5632–5637 (2011).
Yoshida, T. MCAT Elements and the TEF-1 family of transcription factors in muscle development and disease. Arterioscler. Thromb. Vasc. Biol. 28, 8–17 (2008).
Junion, G., Spivakov, M., Girardot, C., Braun, M., Gustafson, E. H., Birney, E. et al. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell 148, 473–486 (2012).
Van den Boogaard, M., Wong, L. Y. E., Tessadori, F., Bakker, M. L., Dreizehnter, L. K., Wakker, V. et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J. Clin. Invest. 122, 2519–2530 (2012).
Mosammaparast, N. & Shi, Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 79, 155–179 (2010).
Steward, M. M., Lee, J.-S., O’Donovan, A., Wyatt, M., Bernstein, B. E. & Shilatifard, A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat. Struct. Mol. Biol. 13, 852–854 (2006).
Stoller, J. Z., Huang, L., Tan, C. C., Huang, F., Zhou, D. D., Yang, J. et al. Ash2l interacts with Tbx1 and is required during early embryogenesis. Exp. Biol. Med. 235, 569–576 (2010).
Lee, Y. F., Nimura, K., Lo, W. N., Saga, K. & Kaneda, Y. Histone H3 lysine 36 methyltransferase Whsc1 promotes the association of Runx2 and p300 in the activation of bone-related genes. PLoS ONE 9, e106661 (2014).
Kim, T., Chen, J., Sadoshima, J. & Lee, Y. Jumonji represses atrial natriuretic factor gene expression by inhibiting transcriptional activities of cardiac transcription factors. Mol. Cell Biol. 24, 10151–10160 (2004).
Li, G., Margueron, R., Ku, M., Chambon, P., Bernstein, B. E. & Reinberg, D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 24, 368–380 (2010).
Wang, Z., Zang, C., Cui, K., Schones, D. E., Barski, A., Peng, W. et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031 (2009).
Murakami, M., Nakagawa, M., Olson, E. N. & Nakagawa, O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt–Oram syndrome. Proc. Natl Acad. Sci. USA 102, 18034 (2005).
Barron, M. R., Belaguli, N. S., Zhang, S. X., Trinh, M., Iyer, D., Merlo, X. et al. Serum Response factor, an enriched cardiac mesoderm obligatory factor, is a downstream gene target for Tbx genes. J. Biol. Chem. 280, 11816–11828 (2005).
Chandrasekaran, S., Peterson, R. E., Mani, S. K., Addy, B., Buchholz, A. L., Xu, L. et al. Histone deacetylases facilitate sodium/calcium exchanger up-regulation in adult cardiomyocytes. FASEB J. 23, 3851–3864 (2009).
Lickert, H., Takeuchi, J. K., von Both, I., Walls, J. R., McAuliffe, F., Adamson, S. L. et al. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature 432, 107–112 (2004).
Dai, Y.-S. p300 functions as a coactivator of transcription factor GATA-4. J. Biol. Chem. 276, 37178–37185 (2001).
Takeuchi, J. K. & Bruneau, B. G. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 459, 708–711 (2009).
Han, P., Hang, C. T., Yang, J. & Chang, C.-P. Chromatin remodeling in cardiovascular development and physiology. Circ. Res. 108, 378–396 (2011).
Davenport, S. L. H., Hefner, M. A. & Mitchell, J. A. The spectrum of clinical features in CHARGE syndrome. Clin. Genet. 29, 298–310 (1986).
Jongmans, M. C. J., Admiraal, R. J., Donk, K. P., van der, Vissers, L. E. L. M., Baas, A. F., Kapusta, L. et al. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J. Med. Genet. 43, 306–314 (2006).
Van Nostrand, J. L., Brady, C. A., Jung, H., Fuentes, D. R., Kozak, M. M., Johnson, T. M. et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 514, 228–232 (2014).
Sano, M., Minamino, T., Toko, H., Miyauchi, H., Orimo, M., Qin, Y. et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 446, 444–448 (2007).
Zhou, P., He, A. & Pu, W. T. in Current Topics in Developmental Biology 100, 143–169 (Elsevier, Philadelphia, PA, USA, 2012).
Frey, N., Katus, H. A., Olson, E. N. & Hill, J. A. Hypertrophy of the heart a new therapeutic target? Circulation 109, 1580–1589 (2004).
He, A., Gu, F., Hu, Y., Ma, Q., Yi, Ye, L., Akiyama, J. A. et al. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat. Commun. 5, 4907 (2014).
Han, P., Li, W., Lin, C.-H., Yang, J., Shang, C., Nurnberg, S. T. et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 514, 102–106 (2014).
Hang, C. T., Yang, J., Han, P., Cheng, H.-L., Shang, C., Ashley, E. et al. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 466, 62–67 (2010).
Wei, J. Q., Shehadeh, L. A., Mitrani, J. M., Pessanha, M., Slepak, T. I., Webster, K. A. et al. Quantitative control of adaptive cardiac hypertrophy by acetyltransferase p300. Circulation 118, 934–946 (2008).
Montgomery, R. L., Davis, C. A., Potthoff, M. J., Haberland, M., Fielitz, J., Qi, X. et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21, 1790–1802 (2007).
Trivedi, C. M., Luo, Y., Yin, Z., Zhang, M., Zhu, W., Wang, T. et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3β activity. Nat. Med. 13, 324–331 (2007).
Zhang, C. L., McKinsey, T. A., Chang, S., Antos, C. L., Hill, J. A. & Olson, E. N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110, 479–488 (2002).
Anand, P., Brown, J. D., Lin, C. Y., Qi, J., Zhang, R., Artero, P. C. et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 154, 569–582 (2013).
Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W. B., Fedorov, O. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Zaidi, S., Choi, M., Wakimoto, H., Ma, L., Jiang, J., Overton, J. D. et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223 (2013).
Smemo, S., Tena, J. J., Kim, K.-H., Gamazon, E. R., Sakabe, N. J., Gómez-Marín, C. et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375 (2014).
Mansour, M. R., Abraham, B. J., Anders, L., Berezovskaya, A., Gutierrez, A., Durbin, A. D. et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377 (2014).
Hu, Z., Shi, Y., Mo, X., Xu, J., Zhao, B., Lin, Y. et al. A genome-wide association study identifies two risk loci for congenital heart malformations in Han Chinese populations. Nat. Genet. 45, 818–821 (2013).
Cordell, H. J., Bentham, J., Topf, A., Zelenika, D., Heath, S., Mamasoula, C. et al. Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat. Genet. 45, 822–824 (2013).
Bergemann, A., Cole, F. & Hirschhorn, K. The etiology of Wolf?Hirschhorn syndrome. Trends Genet. 21, 188–195 (2005).
Glessner, J. T., Bick, A. G., Ito, K., Homsy, J. G., Rodriguez-Murillo, L., Fromer, M. et al. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ. Res. 115, 884–896 (2014).
Vanlerberghe, C., Petit, F., Malan, V., Vincent-Delorme, C., Bouquillon, S., Boute, O. et al. 15q11.2 microdeletion (BP1–BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur. J. Med. Genet. 58, 140–147 (2015).
De Rubeis, S., Pasciuto, E., Li, K. W., Fernández, E., Di Marino, D., Buzzi, A. et al. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 79, 1169–1182 (2013).
Ye, M., Coldren, C., Liang, X., Mattina, T., Goldmuntz, E., Benson, D. W. et al. Deletion of ETS-1, a gene in the Jacobsen syndrome critical region, causes ventricular septal defects and abnormal ventricular morphology in mice. Hum. Mol. Genet. 19, 648–656 (2010).
Turner, J. & Crossley, M. The CtBP family: enigmatic and enzymatic transcriptional co-repressors. Bioessays 23, 683–690 (2001).
Hildebrand, J. D. & Soriano, P. Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol. Cell. Biol. 22, 5296–5307 (2002).
Stefanovic, S., Barnett, P., van Duijvenboden, K., Weber, D., Gessler, M. & Christoffels, V. M. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nat. Commun. 5, 3680 (2014).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Wilbanks, E. G. & Facciotti, M. T. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS ONE 5, e11471 (2010).
Marzluff, W. F., Wagner, E. J. & Duronio, R. J. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat. Rev. Genet. 9, 843–854 (2008).
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Hardcastle, T. J. & Kelly, K. A. baySeq: Empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics 11, 422 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Acknowledgements
This work is supported by Grant-in-Aid for Scientific Research on Innovative Areas, and Grants from the Japan Heart Foundation, the Kanae Foundation for the Promotion of Medical Science, the Takeda Science Foundation and the NIH Fellows Editorial Board to KN.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Nimura, K., Kaneda, Y. Elucidating the mechanisms of transcription regulation during heart development by next-generation sequencing. J Hum Genet 61, 5–12 (2016). https://doi.org/10.1038/jhg.2015.84
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.84
