
Abstract
Uterine leiomyoma (UL) are benign neoplasms arising from the smooth muscle cells of the uterus. One of the established risk factors for UL is African American ethnicity. Studies have consistently shown that African Americans have two to three times higher risk compared with that of non-Hispanic Whites. However, there is still no adequate explanation for the higher risk among African Americans. To investigate the genetic contribution to the observed difference between the African American and European American populations, we conducted an admixture scan in 525 eligible African American women participants to the NIEHS uterine fibroid study (NIEHS-UFS). In models with no stratification, we found multiple genomic regions showing significant and suggestive evidence of association, with chromosomal band 2q32.2 at rs256552 showing the highest score (Z-score=7.86, Bonferroni adjusted P-value=5.5 × 10−12) consistent with the suggestive evidence reported for this genomic region in the Black Women’s Health Study. However, in models stratified by the body mass index (BMI) covariate, chromosome 1q42.2 was the sole genomic region that consistently showed suggestive associations across the BMI categories tested (Z-scores ⩽−3.96, Bonferroni adjusted P-values ⩽0.107). In age-stratified models, a significant association was observed in the older category (age >40) reaching a Z-score of 6.44 (Bonferroni-adjusted P-value=1.64 × 10−7) at rs256552. The mean percentage of European ancestry among cases was lower than that among controls in the NIEHS-UFS study. However, our study did not show a significant association between mean percentage of European ancestry and UL.
Similar content being viewed by others


Introduction
Uterine leiomyoma (UL), or fibroids, are benign neoplasms arising from the smooth muscle cells of the uterus. It is believed that these tumors develop in the majority of American women by the time they reach menopause and become symptomatic in ~25%.1 Despite their benign nature, UL are responsible for significant gynecologic morbidities including excessive bleeding, pelvic pain, urinary incontinence, infertility and pregnancy complications.2, 3 Cumulative exposure to estrogen is believed to be a major etiologic factor,4 and factors that may influence the hormonal milieu, such as obesity, are also believed to be associated with the risk.5 However, the clearly established non-genetic risk factors are age (increasing risk with increasing premenopausal age), menopause (risk decreases with menopause) and African American ethnicity (higher risk compared with that of non-Hispanic Whites).6 There is still no adequate explanation for the higher risk among African Americans. The estimated cumulative incidence remained significantly higher for African Americans after controlling for parity and body mass index (BMI) in an ultrasound screening study.6
The genetic basis of susceptibility to UL is not well understood. Compelling evidence for a genetic liability in the development of these tumors have been demonstrated in familial aggregation studies and twin studies;7, 8, 9, 10 however, findings from genome-wide genome scans (including linkage and admixture mapping scans) and genome-wide association studies reported to date11, 12, 13, 14 have not converged on a plausible mechanism of pathogenesis. Epidemiologic studies of UL in American women enrolled in the Right From The Start cohort and the BioVU DNA repository replicated the results of genome-wide association studies in a Japanese population for two out of the three most significantly associated genes encoding BET1L (blocked early in transport 1 homolog) and TNRC6B (trinucleotide repeat containing 6B).15, 16 Recent studies in NIEHS-UFS (National Institute of Environmental and Health Sciences-Uterine Fibroid Study) has implicated gene variants encoding components of the extracellular matrix17 and replicated the association with TNRC6B.
To investigate whether the African American ethnicity confers a higher risk for UL, we performed admixture mapping in a sample of 393 UL cases of African American descent enrolled in NIEHS-UFS. Admixture mapping provides an efficient way to identify genetic risk variants that underlie the differences in population disease prevalence.18, 19 The idea behind admixture mapping is simple: genetic risk factors for diseases will be more frequent in chromosome regions derived from the ancestral population with the higher disease rate. For recently admixed populations, such as African Americans, admixture-generated linkage disequilibrium extends many millions of base pairs.19 Therefore, only a few thousands of ancestry-informative markers that differ in frequency between the parental populations of the admixed population are sufficient to carry out admixture mapping studies.18 Recently, with the availability of high-density admixture marker maps for several admixed populations, including African Americans and Mexican Americans18, 20 and the development of efficient statistical and computational methods for admixture mapping,19, 21, 22, 23, 24, 25, 26 this approach has drawn considerable attention and successfully identified multiple genetic risk variants for human complex traits (for example, hypertension,27, 28 multiple sclerosis29 and so on). With an additional set of 132 clinically ascertained UL-free controls of African American descent enrolled in NIEHS-UFS, we also estimated the percentage of European ancestry for each case and control and investigated if there was an association between mean percentage of European ancestry and UL.
Despite the success of admixture mapping in gene disease mapping, only one study has used this approach to identify genetic risk variants for UL.13 Wise et al.13 carried out a genome-wide admixture mapping using DNA samples from 2453 UL cases and 2102 controls in the Black Women’s Health Study (BWHS). The mean percentage of European ancestry was found to be significantly associated with the risk for UL in BWHS and genomic regions at chromosomes 2q32-33, 4p16 and 10q26 showed suggestive evidence for association with UL; however, these findings did not overlap those of recent genome-wide association studies in the Japanese 12 and North American14 populations. Our study did not show a significant association between mean percentage of European ancestry and UL. After stratifying by the BMI, our admixture mapping showed suggestive-to-significant associations at chromosome 1q42.2.
Materials and methods
Study population
Detailed characteristics of the study population have been reported;30, 31 only those relevant to the present study will be described. Briefly, a random sample of women, aged 35–51 years, was selected from a computerized list of members of a prepaid urban health plan for enrollment in the NIEHS-UFS.6 Demographic data were collected by self-administered questionnaire. Reproductive and gynecologic history data were collected during a telephonic interview. About 92% of the study population was self-identified as African Americans or non-Hispanic European Americans. Of the enrolled women who were premenopausal and were screened for UL (n=1119), 1045 (93%) had available DNA specimens and among these 574 (54.9%) self-identified as African Americans. The NIEHS-UFS and the present sub-study were approved by the Human Subject’s Review boards at the NIEHS, George Washington University and University of Alabama at Birmingham, respectively. Participants gave written informed consents.
We used Discriminant Analysis of Principal Components32 of genetic data generated from a set of 2682 single-nucleotide polymorphism (SNPs) that were common to our data and to the majority of the reference populations of the International HapMapIII Project to assess population membership for each of the 558 eligible NIEHS-UFS individuals (417 cases and 141 controls, the only eligible AA controls) described in a previous report.31
Covariates
The covariates included age, parity after age 25 (earlier births were not significantly related to fibroid development in the NIEHS-UFS),33, 34 BMI and physical activity.30
Ascertainment
Fibroid status was assessed by ultrasound screening at baseline or by medical record review in 80% of the participants. For those women who had a pelvic ultrasound examination recently at the health plan, the radiology records from that examination were used to assess fibroid status. The remaining premenopausal participants were asked to have a pelvic ultrasound examination at the primary care site. Women for whom neither ultrasound nor medical record review could be conducted were excluded from the analyses. Both a transabdominal and a transvaginal ultrasound examination were performed. The abdominal portion evaluated fibroid change arising from the upper uterus that would not be readily seen with the transvaginal approach alone.
Ancestry informative markers and quality control
Blood DNA was available for 558 eligible participants. Illumina (San Diego, CA, USA) iSelect assay was used to genotype the selected SNPs at the Hudson Alpha Institute for Biotechnology, Huntsville, AL, USA as described.31 We selected and genotyped 1583 MALD (mapping by admixture linkage disequilibrium) SNPs. In selecting this subset of SNPs from the published set of 3011 MALD SNPs,18 we prioritized those SNPs overlapping the candidate regions (at chromosomes 2q37, 3p21, 5p13, 10p11, 11p15, 12q14 and 17q25) identified via linkage scan in the affected sister study,14 or by admixture mapping (2q32-33, 4p16 and 10q26) in BWHS.13 We extended this selected set with MALD SNPs from randomly selected chromosomes (chromosomes 1, 15 and 21) that we were able to include in the iSelect assay.
Admixture mapping
We used ADMIXMAP and assessed individual ancestry proportions to scan the genomic regions of African ancestry that differed significantly from the genome average.23 ADMIXMAP (http://homepages.ed.ac.uk/pmckeigu/admixmap/) is a free program that implements a Markov chain Monte Carlo algorithm for modeling admixture using multilocus genotype data and individuals from admixed populations.19 The applications of ADMIXMAP include inferring distribution of individual admixture proportions and identifying genetic risk variants by admixture mapping. The genetic markers used in ADMIXMAP have been chosen to have different frequencies between the parental populations of the admixed African American population under study.19 The program was downloaded and run using 1000 burn-in iterations and 9000 follow-on iterations and outputting sampled values in every 25 iterations. The allele frequency of each MALD SNP in African and European populations was calculated using allele counts provided in the Supplementary Table of Smith et al.,18 and used as input for ADMIXMAP.18 The HapMap Phase II recombination rate on GRCh37 was used to calculate the genetic distance between the eligible MALD SNPs. These recombination rates are the liftover of HapMap phase II genetic map from human genome build b35 to GRCh37 and can be downloaded from http://hapmap.ncbi.nlm.nih.gov/. The score test implemented in ADMIXMAP was used to test for association between UL and each locus by comparing the observed and expected ancestry proportions at the studied locus in cases only. This test also is similar to that used in Wise et al.13 Statistical significance was assessed using Z-scores and Bonferroni adjusted P-values. A negative Z-score indicates that lower European ancestry at a particular locus compared with the genome-wide average is associated with higher UL risk, whereas a positive Z-score indicates that higher European ancestry is associated with higher UL risk. In Wise et al.,13 a |Z-score|⩾3 indicated suggestive evidence, while a |Z-score|⩾4 indicated a statistically significant result. Here, a |Z-score|⩾3 indicates a suggestive result while a Bonferroni-adjusted P-value⩽0.05 which corresponds to a |Z-score|⩾4.133 indicates a statistically significant result. Since BMI is a confounding factor and ADMIXMAP does not allow for covariates to be modeled, we also performed BMI-stratified analyses in ADMIXMAP (BMI⩽25, BMI>25 and BMI⩽30, BMI>30 and BMI⩽35 or BMI>35).
To test the association between the European heritage and the risk of UL, we estimated the proportions of ancestral populations for each individual and used logistic regression with or without age as a covariate to estimate the odds ratio and its 95% confidence interval. Logistic regression models were also used to perform subgroup analyses based on age (age⩽40 or age>40), parity, BMI (BMI⩽25, BMI>25 and BMI⩽30, BMI>30 and BMI⩽35 or BMI>35), or physical activity (low, moderate, high or very high).
Results
Thirty-three of the 558 typed individuals did not pass the genotyping quality control filter (call rate >90%); this resulted in a study population of 525 individuals (393 UL cases and 132 controls) of African American descent available for the analysis.
The characteristics of the 525 African American participants available for admixture mapping are summarized in Table 1. Mean age was 42.5 for UL cases and 40.5 for controls. The mean age of the studied participants in the cross-sectional NIEHS study differed from the mean age at diagnosis in the prospective BWHS, which was 38.3 and 42.9 years for cases and controls, respectively. Because age is an established risk factor (increased risk with increasing premenopausal age), only age-adjusted results will be compared between the two studies. About 47.6% of cases (187 out of 393) and 54.6% (72 out of 132) of controls had given birth after age 25. The mean BMI was 30.9 for UL cases and 29.5 for controls. Among the UL cases, 45.8% had BMI>30, while only 37.9% controls had BMI>30. Also 29.4% of UL cases and 41.2% control had high or very-high physical activity. Although the mean BMI difference between cases and controls was not significant, admixture mapping models with and without stratification by the BMI covariate were tested because (i) admixture mapping was conducted in a case-only design and (ii) both BWHS35 and NIEHS-UFS6 reported an inverse J-shaped association (non-dose response) between BMI and UL. Also, even though we conducted admixture mapping in a case-only design, models with and without stratification by age were tested to allow comparisons of our results with those reported in BWHS.13
Marker quality control
Of the 1583 SNPs selected for typing, 182 (11.4%) were excluded because the assay failed, the SNP was monomorphic or rare, or the SNP deviated from Hardy–Weinberg equilibrium in the controls; this yielded 1401 MALD SNPs available for analyses. Of note, a large number of the MALD SNPs that we have selected for this study are positional markers spanning the linkage and admixture peaks and flanking regions. The other non-positional MALD SNPs were from a random selection of chromosomes.
Global ancestry analysis
The mean percentage of European ancestry obtained from ADMIXMAP for UL cases and controls were 20.7 and 21.3% (Table 2), respectively. The mean differences with and without adjustment for age from the logistic regression were −1.16 and −1.27%, respectively. Subgroup analysis of global ancestry showed that the mean percentage of European ancestry was lower among UL cases than controls in most comparisons. Although the mean percentage of ancestry among UL cases and controls obtained from our study were similar to those from Wise et al.,13 our study failed to show evidence for significant association between percentage of European ancestry and UL.
Admixture mapping analysis
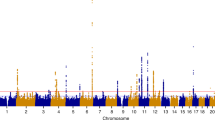
The detailed results of the admixture mapping scan obtained from ADMIXMAP in a case-only design are shown in Supplementary Table S1. For each statistically significant or suggestive peak, the SNP with the highest Z-score is listed in Supplementary Table S2. We have found multiple statistically significant or suggestive peaks at chromosomes 1, 2, 3, 4, 5, 11, 12 and 17 in models with no control for BMI. However, in models stratified by the BMI covariate, most statistically significant or suggestive peaks disappeared. From the quantile–quantile plot (Q–Q plot) of the Z-scores obtained from ADMIXMAP using all 393 cases (Supplementary Figure S1), we observed an inflation of the test statistic at the top of the plot. The inflations of the test statistic can be explained by the confounding effect of BMI since no such inflations were observed in the Q–Q plots for models stratified by BMI (Supplementary Figure S2). Therefore, the models with BMI stratification are used for the interpretation of our study results. Furthermore, using discriminant analysis of principal components, we show that the entire case and control sample used in this study clusters only with the HapMap III reference populations of the African ancestry Yoruba in Ibadan, Nigeria (YRI), Luhya in Webuye, Kenya (LWK) and Maasai in Kinyawa, Kenya (MKK) or African ancestry in Southwest USA (ASW) and not with any other population tested, thus indicating that spurious results due to population structure are unlikely (Supplementary Figure S3).
Chromosome 1q42.2 was the sole genomic region that showed suggestive-to-significant associations at rs7546784 (−6.81⩽Z-scores⩽−3.96, Bonferroni-adjusted 1.40 × 10−8⩽P-value⩽0.107) consistently across the four BMI categories and age categories (Table 3). In agreement with African ancestry being at risk, the association indicates that lower European ancestry at chromosome 1q42.2 increases the risk for UL. It is not clear if this region of chromosome 1q42.2 was screened in BWHS since that study too has not used the full set of available MALD SNPs.18 The signal on chromosome 2q32.2 peaked at rs256552 around position 190.7 Mb with a Z-score of 7.86 and Bonferroni-adjusted P-value of 5.5 × 10−12 in models with no control for BMI. Although the association was not significant across the four BMI categories, suggestive evidence of association (Z-score of 3.04 in the BMI category 30 to 35 and Z-scores around 2 for the other BMI categories) was observed. In age-stratified models, a significant association was observed in the older category (age >40) reaching a Z-score of 6.44 (Bonferroni-adjusted P-value =1.64 × 10−7).
We noticed that candidate SNPs rs7546784 and rs256552 were both common SNPs in African Americans (Table 4). Further re-evaluation of the association by logistic regression with adjustment for BMI and age showed that only rs7546784 was associated with UL (Table 4). Removing the age and BMI from the logistic regression did not affect significantly the results (Supplementary Table S3). Finally, except for the chromosome 1q42.2 signal, no linear relationship was observed between the Z-score and BMI. Changes to the Z-score sign across the BMI categories were observed in only a few instances (at rs10473043 on chromosomal band 5p13.2 and at rs1894223 on chromosomal band 11q24.2).
Discussion
We performed an admixture mapping study of UL in 393 African American UL cases using 1401 ancestry informative SNPs. To our knowledge, this is the second report of admixture mapping for UL after that of BWHS.13 Although we have found several statistically significant peaks on chromosomes 1, 2, 3, 4, 5, 11, 12, 15 and 17, after controlling for the BMI or the age covariate, a sub-region of chromosome 1q42.2 tagged by rs7546784 was the sole region that consistently showed significant associations across the BMI or age categories. The negative Z-score observed at chromosome 1q42.2 is consistent with African ancestry being at higher risk.36 To our knowledge, this locus was not highlighted in any previous genome-wide association studies or admixture study of UL. However, in models with no control for BMI, the strongest association peaked at chromosome 2q32.2 at a location close to the peak reported in BWHS for rs2271767 (Z-score=3.69) at position 192.3.13 Using ADMIXMAP, Wise et al. have found a suggestive evidence for a 2q32-q33 region around rs7573626 (physical position=206.3; Z-score=3.42 using all UL cases), rs2271767 (physical position =192.3, Z-score=3.69 using cases aged <35 years), rs6710083 (physical position=192.3, Z-score=4.10 using surgical cases) and rs920249 (physical position=201.3, Z-score=5.10 using surgical cases aged<35 years).
We were unable to replicate the significant association between the mean percentage of European ancestry and UL reported in BWHS. As indicated by the results shown in Table 2, the mean percentage of European ancestry among cases was lower than that among controls in the NIEHS study, which is in the same direction to that reported in BWHS. However; the mean difference in the NIEHS (means of European ancestry of 20.7 and 21.3% in cases and controls, respectively) was smaller than that in the BWHS (means of 20 and 21.6% for cases and controls, respectively). This smaller mean difference, together with the smaller size of our study sample compared to that of BWHS, account for the lack of statistical significance in our study.
The comparison of the results between the two studies should be interpreted with cautions because: (i) all the participants to NIEHS-UFS were more than 35 years of age at enrollment and only the mean percentage of European ancestry was reported for this age category in BWHS; (ii) contrasting with our study which tested 1401 SNPs on a smaller sample of 525 individuals (393 cases and 132 controls), the data from the BWHS were based on the 30 most informative MALD SNPs37 typed in a larger sample (2453 cases and 2102 controls); and (iii) NIEHS-UFS ascertained clinically the enrolled controls in a cross-sectional study whereas BWHS is a prospective cohort study that assessed the UL affection status in the controls through a questionnaire at the end of the follow-up period. It is clear; however, that given the estimated effect size of 0.086, our study based on 393 cases and 132 controls had limited power to detect difference in mean percentage of European ancestry.
Most of the MALD SNPs selected for the present study map within candidate genomic regions identified by admixture mapping in BWHS or by linkage scan in the affected sister study;14 thus, back replication of the present findings in these discovery studies is an important step toward their validation.
The interpretation of our results from the models with and without BMI stratification and their comparison with those of BWHS remains difficult. First, to enable comparisons between the two studies, we conducted stratified analyses based on the BMI categories used in BWHS; however, the association between BMI and UL peaked in the BMI range 30–34 in NIEHS-UFS30 whereas in BWHS, the peak was observed in the BMI range 25–29.35 Second, as a proxy measure for the steroid-producing body fat, BMI may affect the association with these hormone-dependent tumors through the causal pathway by decreasing the serum level of sex hormone-binding globulin and consequent increase in bioavailability of free steroids,38 through pleiotropy or through linkage disequilibrium between a UL and an obesity genes.39 The interpretation of these results depends on the validity of these alternative etiologic models. If BMI is a risk factor operating in the causal pathway then the data obtained from models with no BMI-stratification are more robust because they are based on the full BMI variance. In the alternative that BMI rather confounds the association between the actual UL genes and the UL outcome, then confidence in the data derived from models with BMI stratification is greater. In either model, the locus on chromosome 1q42.2 remains an attractive candidate.
It is still early to draw conclusions about potential candidate UL genes. The SNP that captured the association at chromosome 1q42.2 is an intronic variant in PCNXL2 (pecanex-like 2 gene), which encodes a transmembrane protein with uncharacterized function. The strongest signal on chromosome 2q32.2 was captured by a variant located in the immediate 3′UTR downstream region of PMS1 (post-meiotic segregation increased 1) whose product belongs to the DNA mismatch repair mutL/hexB family. This locus and the surrounding loci within a 1–2 Mb interval from position 190.74 Mb on chromosome 2q, which is the average physical distance of the admixture mapping resolution, are target candidate loci for follow-up fine mapping. Importantly, with the suggestive association (Z-score=3.69) reported in BWHS at position 192.3 Mb, this is the sole candidate region that we were able to replicate, albeit with moderate association strength, in NIEHS-UFS.
References
Cramer, S. F. & Patel, A. The frequency of uterine leiomyomas. Am. J. Clin. Pathol. 94, 435–438 (1990).
Stewart, E. A. Uterine fibroids. Lancet 357, 293–298 (2001).
Walker, C. L. & Stewart, E. A. Uterine fibroids: the elephant in the room. Science 308, 1589–1592 (2005).
Andersen, J. Growth factors and cytokines in uterine leiomyomas. Semin. Reprod. Endocrinol. 14, 269–282 (1996).
Schwartz, S. M., Marshall, L. M. & Baird, D. D. Epidemiologic contributions to understanding the etiology of uterine leiomyomata. Environ. Health Perspect. 108 (suppl 5), 821–827 (2000).
Baird, D. D., Dunson, D. B., Hill, M. C., Cousins, D. & Schectman, J. M. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am. J. Obstet. Gynecol. 188, 100–107 (2003).
Kurbanova, M., Koroleva, A. G. & Sergeev, A. S. Genetic analysis of the predisposition to uterine myoma. Prevalence and morbidity. Genetika 25, 1122–1124 (1989).
Vikhlyaeva, E. M., Khodzhaeva, Z. S. & Fantschenko, N. D. Familial predisposition to uterine leiomyomas. Int. J. Gynaecol. Obstet. 51, 127–131 (1995).
Treloar, S. A., Martin, N. G., Dennerstein, L., Raphael, B. & Heath, A. C. Pathways to hysterectomy: insights from longitudinal twin research. Am. J. Obstet. Gynecol. 167, 82–88 (1992).
Luoto, R., Kaprio, J., Rutanen, E. M., Taipale, P., Perola, M. & Koskenvuo, M. Heritability and risk factors of uterine fibroids–the Finnish Twin Cohort study. Maturitas 37, 15–26 (2000).
Gross, K. L., Panhuysen, C. I., Kleinman, M. S., Goldhammer, H., Jones, E. S. & Nassery, N. et al. Involvement of fumarate hydratase in nonsyndromic uterine leiomyomas: genetic linkage analysis and FISH studies. Genes Chromosomes Cancer 41, 183–190 (2004).
Cha, P. C., Takahashi, A., Hosono, N., Low, S. K., Kamatani, N. & Kubo, M. et al. A genome-wide association study identifies three loci associated with susceptibility to uterine fibroids. Nat. Genet. 43, 447–450 (2011).
Wise, L. A., Ruiz-Narvaez, E. A., Palmer, J. R., Cozier, Y. C., Tandon, A., Patterson, N. et al. African ancestry and genetic risk for uterine leiomyomata. Am. J. Epidemiol. 176, 1159–1168 (2012).
Eggert, S. L., Huyck, K. L., Somasundaram, P., Kavalla, R., Stewart, E. A., Lu, A. T. et al. Genome-wide linkage and association analyses implicate FASN in predisposition to uterine leiomyomata. Am. J. Hum. Genet. 91, 621–628 (2012).
Edwards, T. L., Michels, K. A., Hartmann, K. E. & Velez Edwards, D. R. BET1L and TNRC6B associate with uterine fibroid risk among European Americans. Hum. Genet. 132, 943–953 (2013).
Edwards, T. L., Hartmann, K. E. & Velez Edwards, D. R. Variants in BET1L and TNRC6B associate with increasing fibroid volume and fibroid type among European Americans. Hum. Genet. 132, 1361–1369 (2013).
Aissani, B., Zhang, K. & Wiener, H. Follow-up to genome-wide linkage and admixture mapping studies implicates components of the extracellular matrix in susceptibility to and size of uterine fibroids. Fertil. Steril. 103, 528–34 e13 (2015).
Smith, M. W., Patterson, N., Lautenberger, J. A., Truelove, A. L., McDonald, G. J., Waliszewska, A. et al. A high-density admixture map for disease gene discovery in African Americans. Am. J. Hum. Genet. 74, 1001–1013 (2004).
Patterson, N., Hattangadi, N., Lane, B., Lohmueller, K. E., Hafler, D. A., Oksenberg, J. R. et al. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 74, 979–1000 (2004).
Collins-Schramm, H. E., Chima, B., Morii, T., Wah, K., Figueroa, Y., Criswell, L. A. et al. Mexican American ancestry-informative markers: examination of population structure and marker characteristics in European Americans, Mexican Americans, Amerindians and Asians. Hum. Genet. 114, 263–271 (2004).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587 (2003).
Montana, G. & Pritchard, J. K. Statistical tests for admixture mapping with case-control and cases-only data. Am. J. Hum. Genet. 75, 771–789 (2004).
Hoggart, C. J., Shriver, M. D., Kittles, R. A., Clayton, D. G. & McKeigue, P. M. Design and analysis of admixture mapping studies. Am. J. Hum. Genet. 74, 965–978 (2004).
Tang, H., Coram, M., Wang, P., Zhu, X. & Risch, N. Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet. 79, 1–12 (2006).
Price, A. L., Tandon, A., Patterson, N., Barnes, K. C., Rafaels, N., Ruczinski, I. et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 5, e1000519 (2009).
Sankararaman, S., Sridhar, S., Kimmel, G. & Halperin, E. Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 82, 290–303 (2008).
Zhu, X. & Cooper, R. S. Admixture mapping provides evidence of association of the VNN1 gene with hypertension. PloS One 2, e1244 (2007).
Rao, W., Ma, Y., Ma, L., Zhao, J., Li, Q., Gu, W. et al. High-resolution whole-genome haplotyping using limited seed data. Nat. Methods 10, 6–7 (2013).
Reich, D., Patterson, N., De Jager, P. L., McDonald, G. J., Waliszewska, A., Tandon, A. et al. A whole-genome admixture scan finds a candidate locus for multiple sclerosis susceptibility. Nat. Genet. 37, 1113–1118 (2005).
Baird, D. D., Dunson, D. B., Hill, M. C., Cousins, D. & Schectman, J. M. Association of physical activity with development of uterine leiomyoma. Am. J. Epidemiol. 165, 157–163 (2007).
Aissani, B., Wiener, H. & Zhang, K. Multiple hits for the association of uterine fibroids on human chromosome 1q43. PloS One 8, e58399 (2013).
Jombart, T., Devillard, S. & Balloux, F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 94 (2010).
Dunson, D. B. & Baird, D. D. A proportional hazards model for incidence and induced remission of disease. Biometrics 58, 71–78 (2002).
Baird, D. D. & Dunson, D. B. Why is parity protective for uterine fibroids? Epidemiology 14, 247–250 (2003).
Wise, L. A., Palmer, J. R., Spiegelman, D., Harlow, B. L., Stewart, E. A., Adams-Campbell, L. L. & Rosenberg, L. Influence of body size and body fat distribution on risk of uterine leiomyomata in U.S. black women. Epidemiology 16, 346–354 (2005).
Baird, D. D., Dixon, D. & Sandler, D. P. African Americans at higher risk than whites for uterine fibroids: ultrasound evidence. Am. J. Epidemiol. 147, S90 (1998).
Ruiz-Narvaez, E. A., Rosenberg, L., Wise, L. A., Reich, D. & Palmer, J. R. Validation of a small set of ancestral informative markers for control of population admixture in African Americans. Am. J. Epidemiol. 173, 587–592 (2011).
Schwartz, S. M. Epidemiology of uterine leiomyomata. Clin. Obstet. Gynecol. 44, 316–326 (2001).
Aissani, B. Confounding by linkage disequilibrium. J. Hum. Genet. 59, 110–115 (2014).
Acknowledgements
We are indebted to the Principal Investigator of the NIEHS uterine fibroid study, Dr Donna Day Baird, for providing access to the NIEHS-UFS resources. The authors would like to express their gratitude to all participants in NIEHS-UFS. We thank Drs Cynthia Morton and Lauren Wise for disclosing the positions of the linkage and admixture peaks before publication of their studies. This study was supported by grant (R01-HD-064398) (to BA) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Zhang, K., Wiener, H. & Aissani, B. Admixture mapping of genetic variants for uterine fibroids. J Hum Genet 60, 533–538 (2015). https://doi.org/10.1038/jhg.2015.60
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.60

{kind=link}