
Abstract
Defects in the mitochondrial translation apparatus can impair energy production in affected tissues and organs. Most components of this apparatus are encoded by nuclear genes, including GFM2, which encodes a mitochondrial ribosome recycling factor. A few patients with mutations in some of these genes have been reported to date. Here, we present two female siblings with arthrogryposis multiplex congenita, optic atrophy and severe mental retardation. The younger sister had a progressive cerebellar atrophy and bilateral neuropathological findings in the brainstem. Although her cerebrospinal fluid (CSF) levels of lactate and pyruvate were not increased, brain magnetic resonance spectroscopy showed a lactate peak. Additionally, her CSF lactate/pyruvate and serum beta-hydroxybutyrate/acetoacetate ratios were high, and levels of oxidative phosphorylation in skin fibroblasts were reduced. We therefore diagnosed Leigh syndrome. Genomic investigation confirmed the presence of compound heterozygous GFM2 mutations (c.206+4A>G and c.2029-1G>A) in both siblings, causing aberrant splicing with premature stop codons (p.Gly50Glufs*4 and p.Ala677Leufs*2, respectively). These findings suggest that GFM2 mutations could be causative of a phenotype of Leigh syndrome with arthrogryposis multiplex congenita.
Similar content being viewed by others


Introduction
Oxidative phosphorylation drives the synthesis of adenosine triphosphate and represents a series of reactions mediated by the mitochondrial respiratory chain that are integral to the inner mitochondrial membrane. Thirteen proteins of the mitochondrial respiratory chain are encoded by mitochondrial DNA and synthesized by the mitochondrial translation machinery, which is coordinated by various nuclear encodes known as mitochondrial translation factors. The process of mitochondrial protein translation consists of four phases: initiation, elongation, termination and ribosome recycling.1, 2 The complementary DNA for factors associated with these phases has been cloned and sequenced in humans. In particular, four elongation factors (EFTu, EFTs, GFM1 and GFM2) have been identified,3, 4, 5, 6 although GFM2 was recently shown to function as a ribosome recycling factor rather than an elongation factor.7
Defects in the mitochondrial translation machinery caused by genetic mutations have been implicated in respiratory deficiency, which is an underlying factor in a number of diseases including Leigh syndrome.8, 9, 10 Only one family with GFM2 defects has been previously described.11 The two patients of this family had microcephaly, a simplified gyral pattern and pachygyria on brain imaging, as well as insulin-dependent diabetes.
We examined a girl with arthrogryposis multiplex congenita, optic atrophy, severe mental retardation, progressive cerebral and cerebellar atrophy and bilateral brainstem neuropathological findings that met the criteria for Leigh syndrome. Novel compound heterozygous mutations in GFM2 were found in both her and her sister, who also presented with arthrogryposis multiplex congenita, optic atrophy and severe mental retardation.
Materials and methods
Patients
Patient II-1 (Figure 1a) was born to nonconsanguineous Japanese parents at 38 weeks of gestation with asphyxia. There was no family history of neuromuscular or neurologic disorders, or congenital malformations. Her body weight was 2104 g (−2.6 s.d.), height 45.0 cm (−1.5 s.d.) and head circumference 31.0 cm (−0.5 s.d.). She had symmetric severe muscle weakness and congenital contractures of the limbs. Her optic disc was slightly pale.

(a) Familial pedigree and mutations. (b) Reverse-transcriptase polymerase chain reaction analysis for the c.206+4A>G mutation using lymphoblastoid cell lines derived from patient II-2, her parents and a control individual. Primers used for the analysis are shown in (c). A single band (296 bp), corresponding to the wild-type allele, was amplified using a control cDNA template. A lower aberrant band was detected from the cDNA of the patient and her mother. (c) Schematic representation of wild type and mutant transcripts determined by sequencing of polymerase chain reaction products. The lower band has a 58 bp deletion of the entire of exon 4, leading to a frameshift (p.Gly50Glufs*41). (d) Schematic representation of mutant transcripts determined by sequencing of polymerase chain reaction products, and primers used for the analysis. The mutant transcript has a deletion of 1 bp in exon 20 and a premature stop codon (p.Ala677Leufs*2). It is likely to undergo nonsense-mediated mRNA decay because the intensity of the mutant allele increased with cycloheximide compared with dimethyl sulfoxide treatment. A full colour version of this figure is available online at the Journal of Human Genetics website.
Brain magnetic resonance imaging showed an enlarged lateral ventricle and hypoplasia of the corpus callosum, but no cortical malformation. Laboratory analyses were normal. Blood lactate and pyruvate were normal (1.8 and 0.1 mM, respectively; lactate/pyruvate ratio, 17.4). She developed episodes of major vomiting and bradycardia. She was initially diagnosed as having dyskinetic cerebral palsy and referred to the division of rehabilitation.
At 3 months, she developed serial spasms that occurred five or six times daily. Electroencephalography showed hypsarrhythmia patterns, which led to a diagnosis of West syndrome. Despite comprehensive treatment including adrenocorticotropic hormone therapy combined with valproic acid, clonazepam, vitamin B6, clobazam and zonisamide, she continued to exhibit frequent subtle tonic seizures. She showed no head control, no ability to roll over, no eye fixation or pursuit and no social smile at 13 months of age. She died at the age of 13 months of dehydration secondary to gastroenteritis. The parents refused a postmortem, but allowed genomic DNA to be extracted from her nails and preserved on the assumption of future investigation for possible genetic disorders.
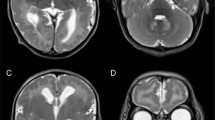
Patient II-2 (Figure 1a), the younger sister of II-1, was born at 39 weeks of gestation without asphyxia 2 years after the death of her older sister. Her body weight was 2500 g (−1.7 s.d.), height 48.2 cm (−0.5 s.d.) and head circumference 32.0 cm (−1.0 s.d.). She was noted to have contractures in all extremities at birth. She was severely hypotonic, and showed poor eye contact and difficulty in feeding. Laboratory analyses revealed normal complete blood counts, liver and kidney function tests, serum electrolytes and creatine kinase levels. Transcranial ultrasonography showed slight atrophy of the cerebellum and brainstem. Brain magnetic resonance imaging revealed abnormal intensity in the subaqueductal area of the midbrain (Figures 2a and b), although the corpus callosum, internal capsule and basal nuclei were spared.

Brain magnetic resonance imaging of patient II-2. A T2-weighted brain magnetic resonance imaging at 1 year of age showed symmetrical high-intensity lesions of the midbrain (a) and slight atrophy of the cerebellum (b). At 7 years of age, the symmetrical high-intensity lesions of the midbrain were unchanged (c), while the diffused cerebellar atrophy was progressive (d).
She developed poorly and remained bedridden and dependent on tube feeding. A tracheotomy was performed at the age of 22 months after repeated respiratory infections, and she became ventilator dependent. She had no epileptic episodes. From 3 years of age, bradycardia appeared and persisted.
At the age of 7, she was referred to our hospital because of severe bradycardia, at ~30 beats min−1. Her body weight was 10.4 g (−3.4 s.d.), height was 93.0 cm (−6.0 s.d.) and her head circumference was 43.5 cm (−5.5 s.d.). Vague eye contact and smiling were noted. Joint contractures in her body, scoliosis and a protruding calcaneus were observed. Deep tendon reflexes were exaggerated, and a positive Babinski sign and ankle clonus were noted. Her optic disks were pale.
Levels of lactate and pyruvate in the blood were normal (1.23 and 0.07 mM, respectively; lactate/pyruvate ratio, 17.6), as were cerebrospinal fluid levels (1.76 and 0.04 mM, respectively; lactate/pyruvate ratio, 44.1). The serum beta-hydroxybutyrate/acetoacetate ratio was 3.9. Other laboratory examinations including blood gases, blood sugar, ammonia, aspartate aminotransferase, alanine aminotransferase, blood urea nitrogen, creatinine, amino acids and urine organic acid analyses were all normal. Electroencephalography showed no abnormalities. The left tibial nerve showed decreased conduction velocity (27.7 m s−1). Electrocardiography revealed sinus bradycardia. Despite extensive treatment including an adrenergic agent, she continued to exhibit frequent bradycardia. Repeated brain magnetic resonance imaging demonstrated a progressive cerebral and cerebellar atrophy (Figures 2c and d). Magnetic resonance spectroscopy showed an intense lactate peak of 1.35 p.p.m. and reduced N-acetyl-aspartate levels (Figure 3).

Magnetic resonance spectroscopy of patient II-2 showing an intense lactate peak at 1.35 p.p.m. and an N-acetyl-aspartate reduction. A full colour version of this figure is available online at the Journal of Human Genetics website.
Determination of enzyme activities
The activities of respiratory chain complexes I, II, III and IV were assayed in mitochondria isolated from the skin fibroblasts of patient II-2, as described previously.12, 13 The activity of each complex was presented as a percentage of the mean value obtained from 12 healthy controls. Together, the percentages of complexes I, II, III and IV activities relative to that of citrate synthase as a mitochondrial enzyme marker or complex II activity were calculated.13
Whole-exome sequencing and Sanger sequencing
Experimental protocols were approved by the institutional review board of Yokohama City University School of Medicine. Written informed consent was obtained from the parents of the two sisters.
Genomic DNA was obtained from peripheral blood leukocytes using QuickGene 610L (Wako, Osaka, Japan) except for patient II-1, in whom DNA was extracted from her nails. Genomic DNA was captured using the SureSelect Human All Exon v5 Kit (51 Mb; Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina HiSeq2000 (Illumina, San Diego, CA, USA) with 101 bp paired-end reads. Exome data processing, variant calling and variant annotation were performed as previously described.14 We filtered out common variants registered in dbSNP135 data, as well as those found in more than five of our 575 in-house control exomes. GFM2 mutations detected by whole-exome sequencing were validated by Sanger sequencing in both sisters and their parents.
Reverse-transcriptase polymerase chain reaction
Lymphoblastoid cell lines were established from patient II-2 and her parents. Total RNA was extracted using the RNeasy Plus Mini kit (Qiagen, Tokyo, Japan) from lymphoblastoid cell lines with or without incubation in 30-μM cycloheximide (Sigma, Tokyo, Japan) for 4 h. Dimethyl sulfoxide was used for vehicle control. A total of 4 μg of total RNA was subjected to reverse transcription using PrimeScript 1st strand synthesis kit with random hexamers (Takara, Otsu, Japan), and 2-μl complementary DNA was used for polymerase chain reaction amplification with the primers listed in Supplementary Table S1. Polymerase chain reaction products were electrophoresed on a 1.5% agarose gel, then the bands were purified using a QIAEXII Gel Extraction Kit (Qiagen) and sequenced.
Results
Enzyme activities
Low complexes III and IV activities were detected in the skin fibroblasts of patient II-2, but there was no change in complexes I and II activities (Table 1).
Whole-exome sequencing and Sanger sequencing
Whole-exome sequencing of patient II-2 and her parents searched for compound heterozygous or homozygous mutations based on an autosomal recessive model. Among four candidate genes consistent with autosomal recessive model, GFM2 was of particular interest because it functions as a ribosome recycling factor. Successive Sanger sequencing of all four family members confirmed the presence of compound heterozygous GFM2 mutations, c.206+4A>G and c.2029-1G>A, in both affected siblings, which had been transmitted from the mother and father, respectively (Figure 1a). Neither mutation was found in the 6500 exomes of the National Heart, Lung, and Blood Institute Exome Sequencing Project Variant Server. c.206+4A>G was not found in any of our in-house 575 Japanese control exomes, while c.2029-1G>A was only detected in one of these.
RNA analysis
Reverse-transcriptase polymerase chain reaction using lymphoblastoid cell lines derived from patient II-2 and her parents clearly demonstrated that the c.206+4A>G mutation caused skipping of GFM2 exon 4, resulting in a smaller amplification band compared with the control (Figures 1b and c). The mutant transcript contained a premature stop codon (p.Gly50Glufs*41), and is likely to undergo nonsense-mediated mRNA decay (NMD) because the intensity of the lower band increased following treatment with cycloheximide, which is an inhibitor of NMD. The c.2125-1G>A mutation caused activation of a cryptic splice acceptor site, leading to the deletion of a single base in exon 20 (Figure 1d). This mutant transcript also had a premature stop codon (p.Ala677Leufs*2) and is also likely to undergo NMD because the intensity of the mutant allele increased with cycloheximide treatment. Therefore it is likely that both transcripts from the two mutant alleles underwent NMD in patient II-2.
Discussion
Hammarsund et al.6 discovered a novel human elongation factor gene, which they designated GFM2, containing 21 exons with conserved exon–intron splice junctions and a complete cDNA sequence of 3033 bp. Its genomic sequence is 45 kb and the 5′-end resides within a CpG island, which is characteristic of a housekeeping gene. The 779 amino acid human GFM2 protein has significant homology to several bacterial elongation factor G proteins. More recently, Tsuboi et al.7 showed that GFM2 did not catalyze translocation but instead mediated ribosome recycling. Indeed, GFM2 is thought to interact with ribosome recycling factor 1 to promote dissociation of ribosomal subunits following the termination of translation. Tsuboi et al. also showed that GFM2 had a significant ribosome-dependent GTPase activity governed by its C-terminal regions, and that GTP hydrolysis appeared to follow the ribosome splitting reaction.
In the current study, we present a girl with Leigh syndrome with multiple congenital arthrogryposis. Whole-exome sequencing and successive Sanger sequencing demonstrated the presence of c.206+4A>G and c.2029-1G>A mutations, which affected transcript splicing leading to premature stop codons: p.Gly50Glufs*41 and p.Ala677Leufs*2, respectively. c.206+4A>G caused a truncating mutation including a GTP-binding site, while c.2029-1G>A caused a deletion of part of domain IV and the entire C-terminal domain. Although functional studies were not performed, these truncations in the GFM2 protein would be expected to affect GTP-binding activity, with a consequent loss of mitochondrial translation efficiency. Additionally, the two aberrant transcripts with premature stop codons appear to undergo NMD in lymphoblastoid cell lines. Therefore, it is likely that both mutations would lead to a loss-of-function of GFM2 at both the mRNA expression and protein function level.
A previous report on GFM2 mutations described very severe phenotypes similar to those of our patient.11 In two siblings with a neurodevelopmental disorder, a homozygous c.2032T>A transversion was identified that resulted in a p.Asp576Glu substitution. This variant occurred in a conserved predicted splice site at the acceptor for exon 17, and was predicted to destroy its function. The patients had microcephaly, a simplified gyral pattern and pachygyria on brain imaging, as well as insulin-dependent diabetes. One of the affected siblings had died, but the cause and age at death were not stated. Moreover, their lactate, pyruvate and oxidative phosphorylation enzyme activities were not investigated, so the level of mitochondrial dysfunction was unknown.
Mutations in other mitochondrial translation factors have also been reported. For instance, a homozygous missense mutation in the mitochondrial elongation factor G1 gene was identified in a patient with fatal neonatal liver failure and lactic acidosis associated with a severe mitochondrial translation defect combined with oxidative phosphorylation deficiency.8 Compound heterozygous nonsense and missense mutations in the same gene were later found in the two sisters with a similar syndrome.9 A homozygous missense mutation in the mitochondrial translation elongation factor Ts has also been reported in two unrelated infants, one affected by mitochondrial encephalomyopathy, the other by fatal hypertrophic cardiomyopathy.10 The latter showed decreased fetal movements, muscular hypotonia and sucking difficulties, which are similar clinical findings to our own cases.
Leigh syndrome is a progressive neurological disease defined by specific neuropathological features associated with lesions in the brainstem and basal ganglia. It may reflect a deficiency of any of the mitochondrial respiratory chain complexes.15, 16 Our patient developed progressive encephalopathy with deteriorating respiratory and psychomotor dysfunction. Magnetic resonance imaging revealed an abnormal intensity in the subaqueductal area of the midbrain with optic and cerebellar atrophy. Although cerebrospinal fluid lactate and pyruvate levels were not increased, an intense lactate peak in the deep white matter was shown in magnetic resonance spectroscopy. Moreover, activities of the oxidative phosphorylation complexes were decreased in skin fibroblasts. Although complexes III and IV activities were not low enough to meet the minor diagnostic criteria for respiratory chain disorders (30–40% activity in a cell line17), these decreased levels might be an evidence of mitochondorial dysfunction caused by GFM2 mutations. Additionally, high lactate/pyruvate and beta-hydroxybutyrate/acetoacetate ratios in the cerebrospinal fluid and serum, respectively, are characteristic of the lesion tissue specificity commonly observed in this syndrome.
The patient was considered to have early-onset Leigh syndrome because of her severe neurological failure with progressive brain atrophy and bilateral necrotizing lesions in the brain stem. Both siblings had similar morphological and symptomatic appearances, but while the older sister had severe epilepsy with infantile spasms, the younger sister was epilepsy free. It is conceivable that adaptive mechanisms act differently in different patients with the same mutation resulting in clinical variability.
Arthrogryposis multiplex congenita results in congenital, nonprogressive contractures in more than two joints and in multiple areas of the body. This has been reported in neonatal mitochondrial disorders resulting from pathology in the central or peripheral nervous system or in muscles.18 Severe bradycardia was also reported in Leigh syndrome.19 Mitochondrial disease caused by GFM2 mutations could be characterized by arthrogryposis multiplex congenita and bradycardia, because the GFM2 protein is highly expressed in skeletal muscle, the heart and fetal liver, which contain a large number of mitochondria.6
We report mutations affecting a nuclear-encoded component of the mitochondrial translation system presenting as Leigh syndrome. Therefore, abnormalities in nuclear genes causing mitochondrial translation defects could represent a novel, potentially broad field of mitochondrial disease. Future research should elucidate the compensatory and regulatory mechanisms at the mitochondrial, cellular and tissue levels that determine the phenotype of mitochondrial disorders caused by mutations in GFM2 and other mitochondrial translation factor genes.
References
Towpik, J. Regulation of mitochondrial translation in yeast. Cell. Mol. Biol. Lett. 10, 571–594 (2005).
Smits, P., Smeitink, J. & van den Heuvel, L. Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J. Biomed. Biotechnol. 2010, 737385 (2010).
Ling, M. F., Merante, F., Chen, H. S., Duff, C., Duncan, A. M. V. & Robinson, B. H. The human mitochondrial elongation factor tu (EF-Tu) gene: cDNA sequence, genomic localization, genomic structure, and identification of a pseudogene. Gene 197, 325–336 (1997).
Xin, H., Woriax, V., Burkhart, W. & Spremulli, L. L. Cloning and expression of mitochondrial translational elongation-factor Ts from bovine and human liver. J. Biol. Chem 270, 17243–17249 (1995).
Gao, J., Yu, L., Zhang, P. Z., Jiang, J. M., Chen, J., Peng, J. S. et al. Cloning and characterization of human and mouse mitochondrial elongation factor G, GFM and Gfm, and mapping of GFM to human chromosome 3q25.1-q26.2. Genomics 74, 109–114 (2001).
Hammarsund, M., Wilson, W., Corcoran, M., Merup, M., Einhorn, S., Grander, D. et al. Identification and characterization of two novel human mitochondrial elongation factor genes, hEFG2 and hEFG1, phylogenetically conserved through evolution. Hum. Genet. 109, 542–550 (2001).
Tsuboi, M., Morita, H., Nozaki, Y., Akama, K., Ueda, T., Ito, K. et al. EF-G2mt Is an Exclusive Recycling Factor in Mammalian Mitochondrial Protein Synthesis. Mol Cell. 35, 502–510 (2009).
Coenen, M. J. H., Antonicka, H., Ugalde, C., Sasarman, F., Rossi, R., Heister, J. et al. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl. J. Med. 351, 2080–2086 (2004).
Antonicka, H., Sasarman, F., Kennaway, N. G. & Shoubridge, E. A. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum. Mol. Genet. 15, 1835–1846 (2006).
Smeitink, J. A. M., Elpeleg, O., Antonicka, H., Diepstra, H., Saada, A., Smits, P. et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am. J. Hum. Genet. 79, 869–877 (2006).
Dixon-Salazar, T. J., Silhavy, J. L., Udpa, N., Schroth, J., Bielas, S., Schaffer, A. E. et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl. Med 4, 138ra78 (2012).
Rahman, S., Blok, R. B., Dahl, H. H. M., Danks, D. M., Kirby, D. M., Chow, C. W. et al. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol 39, 343–351 (1996).
Kirby, D. M., Crawford, M., Cleary, M. A., Dahl, H. H. M., Dennett, X. & Thorburn, D. R. Respiratory chain complex I deficiency – an underdiagnosed energy generation disorder. Neurology 52, 1255–1264 (1999).
Saitsu, H., Nishimura, T., Muramatsu, K., Kodera, H., Kumada, S., Sugai, K. et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 45, 445–449 (2013).
Farina, L., Chiapparini, L., Uziel, G., Bugiani, M., Zeviani, M. & Savoiardo, M. MR findings in Leigh syndrome with COX deficiency and SURF-1 mutations. Am. J. Neuroradiol 23, 1095–1100 (2002).
Finsterer, J. Leigh and Leigh-like syndrome in children and adults. Pediatr Neurol. 39, 223–235 (2008).
Bernier, F. P., Boneh, A., Dennett, X., Chow, C. W., Cleary, M. A. & Thorburn, D. R. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 59, 1406–1411 (2002).
Tulinius, M. & Oldfors, A. Neonatal muscular manifestations in mitochondrial disorders. Semin. Fetal Neonatal Med 16, 229–235 (2011).
Berardo, A., Musumeci, O. & Toscano, A Cardiological manifestations of mitochondrial respiratory chain disorders. Acta Myol 30, 9–15 (2011).
Acknowledgements
We thank Dr Peter Olley (Professor Emeritus, University of Alberta) for English language advice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Fukumura, S., Ohba, C., Watanabe, T. et al. Compound heterozygous GFM2 mutations with Leigh syndrome complicated by arthrogryposis multiplex congenita. J Hum Genet 60, 509–513 (2015). https://doi.org/10.1038/jhg.2015.57
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.57
