
Abstract
We report on a Brachydactyly Type C (BDC) patient with clinically inconspicuous parents. Molecular genetic analyses revealed compound heterozygosity for two GDF5 variants. The variant c.956G>T (p.Gly319Val) was inherited from her mother and has been reported in exome sequencing projects, whereas c.1073T>C (p.Ile358Thr) has never been reported so far. In silico, both variants were predicted to be ‘disease-causing’, but the fact that p.Ile358Thr was predicted by SIFT to be ‘tolerated’ raised our suspicion. Therefore, we performed in vitro assays. To our surprise, GDF5G319V showed pronounced loss of function in luciferase reporter assays and in vitro chondrogenesis, whereas GDF5I358T and GDF5WT had comparable biological activities. Western blot analyses revealed decreased protein levels after overexpression of GDF5G319V. In absence of linkage or de novo mutation, several scenarios could explain the underlying mechanism of the patient’s phenotype. Owing to reduced activity of GDF5G319V in our functional assays, p.Gly319Val might be causative for BDC, but typically evoke an unrecognizably mild phenotype or even nonpenetrance. Another possibility is that our assays failed to pinpoint the disease-causing mechanism of the p.Ile358Thr allele. A final possibility is that compound heterozygosity for p.Ile358Thr and p.Gly319Val is more deleterious to GDF5 activity than either variant alone. Until all possible explanations can be rigorously tested experimentally, a precise recurrence risk counseling for the parents and the affected child is not possible.
Similar content being viewed by others


Introduction
Brachydactylies are a related group of conditions characterized by shortening or absence of skeletal elements in the limbs. They were classified by Bell into different types (A–E) depending on the affected bones.1 Several of them are caused by mutations in genes that encode components of the Bone Morphogenetic Protein (BMP) pathway such as brachydactyly type A1C (BDA1C, MIM 615072), brachydactyly type A2 (BDA2, MIM 112600), brachydactyly type B2 (BDB2, MIM 611377), and brachydactyly type C (BDC, MIM 113100). Characteristic clinical features of BDC include proximally-set thumbs, hyperphalangy of the index finger, shortening of index and middle fingers, and clinodactyly of the fifth finger as well as broad big toes and brachydactyly of the toes.2
Up to now Growth and Differentiation Factor 5 (GDF5, MIM 601146) is the only gene known to be associated with BDC. GDF5 belongs to the BMP subgroup of the TGFβ superfamily and has a crucial role during limb development.3 It is translated as an inactive preproprotein whose signal peptide is removed upon entrance into the secretory pathway. The proprotein is further processed proteolytically by prohormone convertases of the furin type, which predominantly act intracellularly and recognize the basic amino-acid motif ‘RRKRR’ at positions 377–381.3 Extracellular cleavage of GDF5 has been reported as well.4 The mature GDF5 homodimer binds to its receptors BMPR1A (MIM 601299) or BMPR1B (MIM 603248), which assemble with a dimeric BMP type II receptor complex to activate downstream signaling, mainly the SMAD dependent pathway (reviewed in Nickel et al.5).
Besides BDC, GDF5 mutations are associated with a number of other autosomal dominant skeletal disorders, including multiple synostoses syndrome 2 (SYNS2, MIM 610017), proximal symphalangism type 1B (SYM1B, MIM 615298) and brachydactyly type A2 (BDA2, MIM 112600). Semidominant inheritance has been reported for brachydactyly type A1C (BDA1C, MIM 615072), while homozygous loss of function mutations cause autosomal recessive entities including acromesomelic dysplasia Hunter–Thompson type (MIM 201250), Chondrodysplasia Grebe type (MIM 200700) and Du Pan syndrome (fibular hypoplasia and complex brachydactyly, MIM 228900).
Typically, BDC is caused by heterozygous loss of function mutations in GDF5 following an autosomal dominant mode of inheritance with one reported exception. In one large kindred with BDC Schwabe et al.6 suggested semidomiant inheritance, because homozygous carriers of the GDF5 mutation c.517A>G (p.Met173Val) displayed a severe phenotype. In addition, nonpenetrance of BDC in heterozygous mutation carriers has been described as very mild skeletal abnormalities in clinically inconspicuous individuals.7, 8
It is still challenging to assess the disease-causing potential of variants that were identified by sequencing analyses, even if the variants are within a gene that is well known to be associated with the observed phenotype. One possibility to further evaluate newly identified sequence variants is segregation analysis, which can be sometimes hampered by the fact that the number of family members available for genetics analyses is limited. Another option is to check whether the observed variants are reported in databases such as dbSNP,9 the Exome Variant Server of the NHLBI GO Exome Sequencing Project,10 and the browser of the Exome Aggregation Consortium.11 The disease-causing potential can also partly be predicted by in silico tools such as MutationTaster2,12 Poly-Phen 2,13 and SIFT.14 Final insights can be obtained from in vitro analyses, which are of course not part of routine genetic diagnostics.
Here we report on the clinical and molecular genetic results obtained in an affected girl with BDC. Our results from state of the art database research, in silico predictions as well as functional in vitro assays failed to unequivocally classify the clinical significance of the GDF5 variants. The respective challenge of such experimental analyses is discussed.
Materials and methods
Clinical investigation and molecular genetic analyses
Clinical investigations followed the provision of the Declaration of Helsinki principles. For genetic analyses informed consent was obtained from the mother who is also the legal guardian of her affected daughter. Genomic DNA was extracted from blood. Sequencing analysis of GDF5 was performed as described previously.6 Identified variants were confirmed using alternative primers (forward 5′-AGGGAATGACTTCTGGGTGTT-3′, reverse 5′-GGAGTTCATCAGGGTCTGGA-3′). To evaluate compound heterozygosity, an amplicon spanning both variants was amplified from the patient’s DNA (primer: forward 5′-AGGGAATGACTTCTGGGTGTT-3′, reverse 5′-GGAGTTCATCAGGGTCTGGA-3′) and, after A-tailing, cloned into the pTA-GFP vector. Inserted fragments obtained from single clones were sequenced as described above. Nucleotide positions are given referring to GenBank entry RefSeq: NM_000557.2. Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence. The initiation codon is codon 1.
Cloning of constructs
The coding sequence of human GDF5WT was amplified from pcDNA3.1 (kindly provided by Frank Luyten) using primers (forward 5′-ATACGTCTCACATGAGACTCCCCAAACTCCT-3′, reverse 5′-TGGATCCCTACCTGCAGCCACACGACT-3′) that contained restriction sites for BsmbI and BamHI and ligated into the pSLAX13 shuttle vector. The missense variants c.956G>T coding for p.Gly319Val and c.1073T>C coding for p.Ile358Thr, respectively, were inserted into the coding sequence of human GDF5WT within pSLAX13 via site-directed mutagenesis (primer: G319V forward 5′-GCTGGAGGCCTGGGAACGGGTCAGGGCCGTGGACCTCCGTG-3′, G319V reverse 5′-CACGGAGGTCCACGGCCCTGACCCGTTCCCAGGCCTCCAGC-3′, I358T forward 5′-GACCTGTTCTTTAATGAGACTAAGGCCCGCTCTGGC-3′, I358T reverse 5′-GCCAGAGCGGGCCTTAGTCTCATTAAAGAACAGGTC-3′) and confirmed by sequencing of the entire insert including the coding sequence of GDF5. For luciferase assays, sequences of GDF5WT, GDF5G319V and GDF5I358T as well as murine Bmpr1a and Bmpr1b 15 were cloned from pSLAX13 into the expression vector pCS2+ using ClaI.
Functional analyses
The luciferase reporter gene assay using NIH/3T3 cells (ATCC) was performed as described previously.16 Cells were transfected with the indicated GDF5 expression plasmids. For virus production the coding sequences of GDF5WT, GDF5G319V and GDF5I358T were cloned into the RCAS(BP)A vector after ClaI restriction. Viruses were produced as described previously.16 Viral titers were determined to ensure the infection of equal amounts for each tested variant. For micromass cultures, mesenchymal progenitor cells were obtained and cultured as described previously.16 Cells were infected with empty RCASBP(A) virus or virus overexpressing GDF5WT, GDF5G319V or GDF5I358T using a viral titer of 0.5 × 107 PFU/ml and seeded as drop cultures into 24-well plates. After 4 days, cultures were stained with Alcian blue (for details see Stange et al.16). For western blots, micromass cultures were infected with empty RCAS(BP)A virus or virus expressing GDF5WT, GDF5G319V or GDF5I358T using a viral titer of 1 × 107 PFU ml−1. Sample preparation and western blot were performed as described previously.16 Owing to different antibody conditions the ACTIN loading was not performed on the same protein gel as the GDF5 detection. However, for both analyses equal protein amounts and sample volumes were applied and run in parallel. The results were reproducible, but for space reasons only one representative experiment of each assay is shown.
Results
Clinical and molecular findings in an affected girl with brachydactyly type C
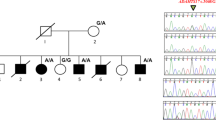
A clinically inconspicuous mother presented her 3-year old daughter because of bilateral hand malformation. The father was reported to be clinically unaffected, but he was not available for physical examination and molecular genetic analyses. The hands and feet of the affected girl show classical features of BDC (Figures 1a–c). Sequencing analyses of GDF5 revealed that the index patient carried two sequence alterations in a heterozygous state. The features of both variants are summarized in Table 1. The nonsynonymous substitution c.956G>T leads to an amino-acid exchange of glycine at position 319 to valine (p.Gly319Val). This variant (hereafter denoted as G319V) was also detected in a heterozygous state in the clinically inconspicuous mother who, unfortunately, rejected further evaluation of her hands using X-rays. The second nonsynonymous substitution, c.1073T>C, leads to an amino-acid exchange of isoleucine at position 358 to threonine (p.Ile358Thr). Theoretically, this variant (hereafter denoted as I358T) could have occurred de novo and be located on either allele or could be inherited from the father who was not available for genetic testing. Compound heterozygosity was confirmed by subcloning of both alleles (Figure 1d, Supplementary Figure S1). Both variants are located in the proregion of GDF5 where G319V affects the core domain, which has been identified in several BMPs and shown to interact with the dimeric, mature domain.17 In contrast, I358T is located between the core domain and the cleavage site that is used to separate the propeptide from the mature protein. As shown in Figure 1e, the amino-acid residues of human GDF5 at positions 319 and 358 are highly conserved in vertebrates. Both variants were predicted to be ‘disease causing’ by MutationTaster2 12 and to be ‘possibly damaging’ by PolyPhen-2.18 Although predicted to be ‘damaging’ by SIFT,14 the sequence variant underlying G319V is listed in dbSNP 9 without frequency data as it has been identified in one European American by the NHLBI Exome Sequencing Project.10 According to the Exome Aggregation Consortium browser it was identified in seven unrelated Europeans.11 In contrast, the amino-acid exchange I358T is predicted to be ‘tolerated’ by SIFT, but the underlying sequence variant has never been described before.

Brachydactyly type C patient carries GDF5 alterations p.Gly319Val and p.Ile358Thr located in the proregion. (a, b) The hands of the 3-year-old girl show classical features of Brachydactyly type C (BDC). Index and middle fingers are shortened. Enlarged and misshaped epiphysis of the index finger is marked by an arrowhead (a). The first metacarpal is shortened leading to a proximally set thumb (asterix); in addition the proximal phalanx of the thumb shows an abnormal shape. Note the clinodactyly of the fifth fingers and the slightly laterally deviated third and fourth fingers of the right hand. (c) At the feet, the patient shows shortened toes and lateral deviation of the broadened great toes. (d) In the index patient, we identified two GDF5 variants, c.956G>T and c.1073T>C, which are predicted to cause the amino-acid exchanges p.Gly319Val and p.Ile358Thr. The mother is clinically inconspicuous, but heterozygous carrier of c.956G>T (p.G319V). Horizontal bars indicate individuals that were available for clinical examination and molecular genetics analyses. (e) Schematic view of the preproprotein of GDF5 encompassing the signal peptide (SP), the proregion and the mature domain. The core domain was identified previously as a subdomain within the proregion.17 Both investigated amino-acid alterations affect the proregion of GDF5, while G319V is located within the core domain. The affected amino acids are conserved in vertebrates (Hs, Homo sapiens; Mm, Mus musculus; Gg, Gallus gallus; Dr, Danio rerio; Xt, Xenopus tropicalis).
Taken together, based on in silico predictions G319V is more likely to be pathogenic, but has been described in reportedly healthy individuals as well as observed in the clinically inconspicuous mother. In contrast, I358T might be tolerated according to one out of three prediction tools, but lacks experimental evidence for phenotypic association. We therefore decided to perform functional validation assays that are well established in our lab for both GDF5 variants.
Functional analyses of the variants GDF5G319V and GDF5I358T
First, we investigated the SMAD-dependent gene induction using a luciferase-based SMAD-binding element reporter.19 GDF5WT is known to activate the SMAD signaling cascade after binding of the mature dimer to its receptors BMPR1A or BMPR1B. As shown in Figure 2a, GDF5WT and GDF5I358T efficiently activated luciferase expression upon transfection with Bmpr1a and Bmpr1b, respectively. Since BMPR1B is the high affinity receptor for GDF5,20, 21 signaling via this receptor was more pronounced than signaling via BMPR1A. In contrast, GDF5G319V showed only little activation of SMAD signaling via BMPR1A and reduced activity upon Bmpr1b transfection. It can thus be concluded that GDF5G319V but not GDF5I358T may constitute a poorly active variant with respect to SMAD signaling.

GDF5G319V but not GDF5I358T shows reduced biological activity. (a) NIH/3T3 cells were transfected with either control vector, GDF5WT, GDF5G319V or GDF5I358T coding vectors as indicated and cotransfected with Bmpr1a or Bmpr1b. Cells were additionally transfected with the luciferase vectors pRL-TK and pGL3ti-SBE. Relative luciferase expression of the control (empty vector) without receptor cotransfection was set to 1. GDF5WT induces SMAD signaling upon receptor cotransfection. Luciferase expression induced by GDF5I358T is comparable to GDF5WT, whereas GDF5G319V shows considerably attenuated SMAD signaling compared to GDF5WT. Bars represent mean±s.d. of triplicates samples from one representative experiment. (b) Micromass cultures were infected with empty control virus or virus expressing GDF5WT, GDF5G319V or GDF5I358T and stained with Alcian blue to visualize extracellular matrix (ECM) of cartilage tissue (n=3). Chondrogenic differentiation is strongly induced by GDF5WT and GDF5I358T. Although GDF5G319V induced ECM formation to some extent, the prochondrogenic activity is clearly diminished in comparison to GDF5WT. Bars represent mean±s.d. of triplicate from one representative experiment. (c) An anti GDF5 antibody was used to perform western blot of supernatants and cell pellets of micromass cultures that were infected with empty control virus or virus expressing GDF5WT, GDF5G319V or GDF5I358T. Proproteins and mature GDF5 can be detected upon expression of GDF5WT in both fractions. The protein amount is considerably reduced when GDF5G319V is expressed, while GDF5I358T shows a similar protein expression pattern as GDF5WT. Samples of the supernatant were loaded onto the same gel, but noncontiguously. Owing to different antibody conditions protein samples for GDF5 detection and the ACTIN loading control were applied on different gels, which were run in parallel. A full color version of this figure is available at the Journal of Human Genetics journal online.
Next, we aimed at exploring the biological activity of both variants. GDF5WT has a well-known prochondrogenic capacity that can be assessed in a chicken limb bud micromass culture-based chondrogenesis assay.22 Limb bud-derived chicken cells were infected with GDF5WT and its variants GDF5G319V and GDF5I358T using the RCAS virus system, which achieves a mild overexpression.23 The accumulation of cartilaginous matrix was visualized and quantified by Alcian blue staining. As shown in Figure 2b, pronounced accumulation of chondrogenic matrix occurred upon GDF5WT expression. The activity of GDF5I358T was again similar to GDF5WT, whereas GDF5G319V induced markedly lower amounts of cartilaginous matrix. Again, GDF5G319V constituted the variant with a compromised prochondrogenic capacity in comparison with GDF5I358T or GDF5WT.
The reduced activation of SMAD signaling and prochondrogenic activity of GDF5G319V may have been caused by impaired biological activity of GDF5G319V or by lower protein levels of this variant. To assess the influence of both GDF5 variants on proprotein and mature protein levels we performed western blot analyses of cell pellets and culture supernatants obtained from chicken micromass cultures expressing the different GDF5 constructs. After infection of GDF5WT both, the different proproteins and the mature protein (~25 kDa) were detected in the cell pellet and as secreted forms in the medium (Figure 2c). The GDF5 levels were comparable after infection of GDF5I358T or GDF5WT. In contrast, infection with the GDF5G319V expression vector yielded only little recombinant protein, which was processed and secreted, despite using the same amount of virus.
Taken together, these experiments indicate that c.956G>T is the more likely disease-causing mutation as it fails to generate similar protein amounts of the encoded GDF5G319V as the wildtype allele, whereas c.1073T>C is likely to cause a presumably mild effect, since the encoded GDF5I358T did not show any abnormalities in the functional assays performed.
Discussion
Here we report on an affected girl with a classical BDC phenotype displaying compound heterozygosity for the GDF5 variants c.956G>T (G319V) and c.1073T>C (I358T). Even after combining database knowledge with in silico predictions and in vitro analyses, the clinical interpretation of the results is inconclusive and a reliable estimation of recurrence risks for future offspring of the mother or the index patient is not possible.
Some GDF5-related skeletal disorders are associated with mutations in other genes. Multiple synostoses syndrome and proximal symphalangism for example can be caused by mutations in NOGGIN (NOG, MIM 602991; SYNS1, MIM 18650; SYM1A, MIM 185800) or in GDF5 (SYNS2; SYM1B), acromesomelic chondrodysplasia Grebe type can be caused by mutations in GDF5 or in BMPR1B24, and BDA2 can be caused by a heterozygous duplication of a regulatory element downstream of BMP2 (MIM 112261) 25 or by mutations in BMPR1B or in GDF5. Although, GDF5 is currently the only gene known to be associated with BDC, one might speculate that mutations in interaction partners, for example in BMPR1B, may cause a similar phenotype. Our analysis is limited in that we did not perform exome sequencing in the patient described here. Thus, we cannot exclude that there might be a causative mutation hidden in a yet unknown gene causing BDC. At least for the GDF5 locus, we used SNPs and copy number analyses (data not shown) to exclude larger deletions that might be combined with the observed variants.
In comparison with classical autosomal dominant disorders with full penetrance, genetics counseling in BDC is more complicated since incomplete penetrance has been repeatedly reported.26, 27 In some studies a more detailed assessment of hand radiographs elucidated mild phenotypic features of BDC in mutation carriers.2, 7, 28, 29 However, the BDC-causing GDF5 point mutation c.206insG showed complete nonpenetrance in some of the investigated individuals.2 Unfortunately, further evaluation of other family members by X-ray analysis was not possible. However, these problems are not uncommon in genetic counseling. Even if X-ray analysis revealed a mild phenotype in the mother, who carries the G319V variant, we might still question whether this variant is a disease-causing mutation, as it was observed in the Exome Aggregation Consortium database with an allele frequency of 0.0001149 in Europeans. This number indicates that roughly 1:30 000 Europeans carry this variant, without being diagnosed with overt BDC. In this collection of sequencing projects, individuals affected by severe pediatric diseases were removed. However, the database may still contain sequencing results from subjects only mildly affected by BDC, whose phenotypes can only be recognized by X-ray analyses. And still, even when we obtained normal X-ray results from these subjects, we still have to consider that the GDF5 mutation c.206insG may show complete nonpenetrance as observed before in some carriers,2 and the same may be true for G319V in the patient’s mother and in the carriers identified by the exome sequencing projects.
Here we provide evidence that GDF5G319V has indeed reduced functionality, whereas GDF5I358T showed normal results in the assays performed. There are two characteristics of the GDF5 variants that support our findings: (1) The amino-acid substitution; in G319V a glycine residue is replaced by a valine. This exchange is predicted to be ‘damaging’ by SIFT. Although both amino acids are nonpolar and neutral, glycine is more flexible than any other amino acid potentially essential for the three dimensional structure. In contrast, the amino-acid exchange in I358T has been predicted to be ‘tolerated’, although the nonpolar neutral residue isoleucine is replaced by the polar neutral residue threonine. We note that SIFT is a multistep procedure that operates with closely related protein sequences that may share similar functions and calculates normalized probabilities for all possible substitutions from the alignment of the chosen sequences. Thus, it takes function, conservation and substitution into account. (2) The position within GDF5; G319V is located in the core fragment of the GDF5 proregion, which is a folded subdomain within the proregions of several BMPs such as GDF5, BMP2 and dpp, the Drosophila ortholog of BMP2.17 Mutations within this core domain have been repeatedly found to be functionally relevant.2 Recently, our group identified two other mutations within the core domain of GDF5, p.T201P and p.L263P, that both showed attenuated SMAD activation and diminished chondrogenic activity associated with reduced protein levels, most probably caused by structural alterations in GDF5 due to the mutations.16, 30 Our findings are in line with these observations, as GDF5G319V displayed reduced functionality and the alteration is located within this core domain, which is not the case for GDF5I358T.
The situation with our index patient is relatively problematic as there is no genetic data available from the father. This limitation leads to unproven assumptions, for example, that I358T may have occurred de novo and thus is likely to be the causative mutation. At least two out of three prediction tools support this hypothesis. Again, we can refer to our in vitro analyses where we obtained normal results for GDF5I358T. Still, we cannot be certain that our assays were the appropriate tests suited to pinpointing the disease-causing mechanism. Even if these assays are well established in our lab, they are limited and it is generally impossible to address all facets of functional GDF5 impairment using in vitro assays. Expression constructs, for example, cannot recapitulate the original genomic environment and do not contain intronic sequences. Thus, cloned variants cannot reliably cover the aspects of transcriptional control and RNA processing.
The penetrance of a mutation can be modulated on several levels. Alterations of the DNA sequence can affect binding of transcription factors. Alterations in the sequence of the primary transcript may interfere with splicing, and differences in the mRNA sequence may influence binding of RNA-binding proteins as well as interactions with microRNAs thereby affecting mRNA stability and translational efficiency. Alterations in the open reading frame may cause an amino-acid exchange which affects protein folding or posttranslational modifications including proteolytic processing or half life. If GDF5I358T levels turned out to be much lower in vivo than levels of GDF5G319V or GDFWT, the effects of GDF5I358T would be less dominant in terms of heterodimer formation and function. Therefore, despite the normal findings in the functional assays reported here, the I358T variant may still be capable of modulating the penetrance of G319V. To assess this issue experimentally, one might consider coexpression or coinfection experiments of both variants in vitro. However, these conditions would still fail to mimic the patient’s situation in vivo. A luciferase reporter gene assay does not show an adequate dose response, as the transfection of the twofold amount of GDF5 expressing plasmid does not necessarily lead to a twofold induction of luciferase activity. For the coinfection of two viral constructs, for example in micromass cultures, one could use different virus subtypes, but still, equal expression levels of RCASBP(A) and RCAS(BP)B cannot be ensured.
In conclusion, our analyses provided some insights into the potential role of the GDF5 variants detected in the index patient. Yet, the definite reason for her phenotype was not identified, and the recurrence risk can’t be reliably estimated. There are four different scenarios possible to explain the patients phenotype: (1) G319V is causative and shows nonpenetrance in the mother; (2) I358T is causative and our in vitro assays failed to identify the underlying pathogenetic mechanism; (3) compound heterozygosity had at least a modifying influence on the G319V allele thus provoking its penetrance in the clearly affected individual presented here; or (4) another (undiagnosed and unidentified) mutation is causative and remained unnoticed by our functional analyses.
References
Bell, J. in Treasury of Human Inheritance (ed Penros, L .) 1–31 (Cambridge Univ. Press, London, UK, 1951).
Everman, D. B., Bartels, C. F., Yang, Y., Yanamandra, N., Goodman, F. R., Mendoza-Londono, J. R. et al. The mutational spectrum of brachydactyly type C. Am. J. Med. Genet. 112, 291–296 (2002).
Luyten, F. P. Cartilage-derived morphogenetic protein-1. Int. J. Biochem. Cell Biol. 29, 1241–1244 (1997).
Ploger, F., Seemann, P., Schmidt-von Kegler, M., Lehmann, K., Seidel, J., Kjaer, K. W. et al. Brachydactyly type A2 associated with a defect in proGDF5 processing. Hum. Mol. Genet. 17, 1222–1233 (2008).
Nickel, J., Sebald, W., Groppe, J. C. & Mueller, T. D. Intricacies of BMP receptor assembly. Cytokine Growth Factor Rev. 20, 367–377 (2009).
Schwabe, G. C., Turkmen, S., Leschik, G., Palanduz, S., Stover, B. & Goecke, T. O. et al. Brachydactyly type C caused by a homozygous missense mutation in the prodomain of CDMP1. Am. J. Med. Genet. A 124A, 356–363 (2004).
Camera, G., Camera, A., Costa, M. & Mantero, R. Pitfalls of genetic counselling in brachydactyly type C. Am. J. Med. Genet. 53, 199–201 (1994).
Savarirayan, R., White, S. M., Goodman, F. R., Graham, J. M. Jr ., Delatycki, M. B., Lachman, R. S. et al. Broad phenotypic spectrum caused by an identical heterozygous CDMP-1 mutation in three unrelated families. Am. J. Med. Genet. A 117A, 136–142 (2003).
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001).
Exome Variant Server (NHLBI GO Exome Sequencing Project (ESP), Seattle, WAURL http://evs.gs.washington.edu/EVS/).
Exome Aggregation Consortium (ExAC), Cambridge, MAURL http://exac.broadinstitute.org.
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Degenkolbe, E., Konig, J., Zimmer, J., Walther, M., Reissner, C., Nickel, J. et al. A GDF5 point mutation strikes twice—causing BDA1 and SYNS2. PLoS Genet. 9, e1003846 (2013).
Stange, K., Thieme, T., Hertel, K., Kuhfahl, S., Janecke, A. R., Piza-Katzer, H. et al. Molecular analysis of two novel missense mutations in the GDF5 proregion that reduce protein activity and are associated with brachydactyly type C. J. Mol. Biol. 426, 3221–3231 (2014).
Kuhfahl, S., Hauburger, A., Thieme, T., Groppe, J., Ihling, C., Tomic, S. et al. Identification of a core domain within the proregion of bone morphogenetic proteins that interacts with the dimeric, mature domain. Biochem. Biophys. Res. Commun. 408, 300–305 (2011).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Jonk, L. J., Itoh, S., Heldin, C. H., ten Dijke, P. & Kruijer, W. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J. Biol. Chem. 273, 21145–21152 (1998).
Heinecke, K., Seher, A., Schmitz, W., Mueller, T. D., Sebald, W. & Nickel, J. Receptor oligomerization and beyond: a case study in bone morphogenetic proteins. BMC Biol. 7, 59 (2009).
Kotzsch, A., Nickel, J., Seher, A., Sebald, W. & Muller, T. D. Crystal structure analysis reveals a spring-loaded latch as molecular mechanism for GDF-5-type I receptor specificity. EMBO J. 28, 937–947 (2009).
Seemann, P., Schwappacher, R., Kjaer, K. W., Krakow, D., Lehmann, K., Dawson, K. et al. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J. Clin. Invest. 115, 2373–2381 (2005).
Ibrahim, D. M., Hansen, P., Rodelsperger, C., Stiege, A. C., Doelken, S. C., Horn, D. et al. Distinct global shifts in genomic binding profiles of limb malformation-associated HOXD13 mutations. Genome Res. 23, 2091–2102 (2013).
Graul-Neumann, L. M., Deichsel, A., Wille, U., Kakar, N., Koll, R., Bassir, C. et al. Homozygous missense and nonsense mutations in BMPR1B cause acromesomelic chondrodysplasia-type Grebe. Eur. J. Hum. Genet. 22, 726–733 (2014).
Dathe, K., Kjaer, K. W., Brehm, A., Meinecke, P., Nurnberg, P., Neto, J. C. et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am. J. Hum. Genet. 84, 483–492 (2009).
Haws, D. V. Inherited brachydactyly and hypoplasia of the bones of the extremities. Ann. Hum. Genet. 26, 201–212 (1963).
Sanz, J. G. S. Type C brachydactyly transmitted through four generations. Ann. Genet. 31, 43–46 (1988).
Galjaard, R. J., van der Ham, L. I., Posch, N. A., Dijkstra, P. F., Oostra, B. A., Hovius, S. E. et al. Differences in complexity of isolated brachydactyly type C cannot be attributed to locus heterogeneity alone. Am. J. Med. Genet. 98, 256–262 (2001).
Robin, N. H., Gunay-Aygun, M., Polinkovsky, A., Warman, M. L. & Morrison, S. Clinical and locus heterogeneity in brachydactyly type C. Am. J. Med. Genet. 68, 369–377 (1997).
Thieme, T., Patzschke, R., Job, F., Liebold, J., Seemann, P., Lilie, H. et al. Biophysical and structural characterization of a folded core domain within the proregion of growth and differentiation factor-5. FEBS J. 281, 4866–4877 (2014).
Stenson, P. D., Mort, M., Ball, E. V., Shaw, K., Phillips, A. & Cooper, D. N. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 133, 1–9 (2014).
Acknowledgements
We thank Johannes Grünhagen and Gundula Leschik for technical assistance and Lutz Schomburg for critical remarks on the manuscript. This work was funded by a doctoral fellowship of the Sonnenfeld-Stiftung to KS. Contributions were made possible by DFG funding through the Berlin-Brandenburg School for Regenerative Therapies GSC 203 (KS) and by DFG funding MU880/11-01 (CEO). All funding sources had no influence on any aspect of the presented study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Stange, K., Ott, CE., Schmidt-von Kegler, M. et al. Brachydactyly Type C patient with compound heterozygosity for p.Gly319Val and p.Ile358Thr variants in the GDF5 proregion: benign variants or mutations?. J Hum Genet 60, 419–425 (2015). https://doi.org/10.1038/jhg.2015.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.48
