
Abstract
The Kra–Dai linguistic family includes Thai and Lao as well as a great number of languages spoken by ethnic minorities in Southeast Asia. In Thailand, a dozen of other Kra–Dai languages are spoken in addition to Thai, the national language. The genetic structure of the Kra–Dai-speaking populations in Thailand has been studied extensively using uniparentally inherited markers. To extend this line of genetic investigation, this study used 15 autosomal microsatellites of 500 individuals from 11 populations, belonging to nine Kra–Dai ethnicities, namely, the Kaleung, Phu Thai, Saek, Nyo, Lao Isan, Yuan, Black Tai, Phuan and Lue. These ethnolinguistic groups are dispersed in three different geographic regions of Thailand, that is, Northern, Northeastern and Central. The results show a very low average of pairwised Fst (0.0099), as well as no population substructure based on STRUCTURE analysis, indicating genetic homogeneity within the Kra–Dai-speaking group, possibly owing to shared linguistic ancestry. The Mantel test, an analysis of molecular variance, and the approximate Bayesian computation procedure employed to evaluate potential factors for driving genetic diversity revealed that language is the predominant factor affecting genetic variations, whereas geography is not. The result of distance-based clustering analyses and spatial analysis of molecular variance revealed genetic distinctions of some populations, reflecting the effects of genetic drift and gene flow on allele frequency within populations, in concordance with the result of R-matrix regression. The genetic and linguistic affiliations of the contemporary Kra–Dai-speaking groups are consistent with each other despite certain deviation due to various evolutionary factors that may have occurred during their migrations and resettlements.
Similar content being viewed by others


Introduction
The Kra–Dai (also known as Tai–Kadai) language family comprises ~95 languages spoken in Southern China and Southeast Asia. In Mainland Southeast Asia, over 100 million people speak ~55 Kra–Dai languages. The languages in this family make up four major subgroups, namely Tai, Kam–Sui, Kra and Hlai. The Tai branch is typically further classified into three sub-branches, namely Central, Northern and Southwestern.1, 2 The most widely spoken Kra–Dai languages are Thai and Lao, which are the official and national languages of the Kingdom of Thailand and the Lao People’s Democratic Republic. Both Thai and Lao belong to the Southwestern grouping.1, 3 Among the estimated 65.9 million people in Thailand, only around 20 million speak Thai as a first language. In contrast, ~40 million people in Thailand use Thai as a second language and speak their own language as their first language in their home. These ethnic languages include different Tai languages, such as Phu Thai, Nyo and Lao, as well as languages from the Austroasiatic, Hmong–Mien, Sino-Tibetan and Austronesian families.4, 5
Thailand lies in the geographical heart of Southeast Asia, and borders Laos to the Northeast, Cambodia to the Southeast, Myanmar to the West and Malaysia to the South. In addition, it is located in close proximity to Vietnam and Southern China to the Northeast and North, respectively. Before the arrival of Tai speakers, various ethnolinguistic groups dominated the territory of present-day Thailand. The Austroasiatic peoples, specifically the Lua’, the Mon and the Khmer, are regarded as the indigenous peoples of present-day Thailand. The Northern region was inhabited by the Lua’ before coming to be dominated by the Mon, who had originally established the ancient Dvaravati kingdom in Central Thailand. In the Northeast, its territory was mainly under the control of the Angkorian Khmer Empire with traces of Dvaravati influence in certain parts of the region.6, 7
At the turn of the second millennium CE, prehistorical Tai-speaking groups started to spread from their homeland in Southeastern China to the area of present-day Thailand.8, 9, 10 In historical times, especially in the 18th century CE, a number of movements and resettlements of Tai-speaking groups occurred continuously owing to diverse reasons including economy, society and military invasion. An example is the forced migration of Yong,11 Lue,12 Shan13 and Khuen speakers6, 14 to Northern Thailand, as well as the mass resettlement of Phuan,15, 16 Black Tai,17 Phu Thai,18 Nyo,19 Kaleung20 and Saek speakers21, 22 in Central and Northeastern regions in the late 18th century.
In recent years, there have been many studies of human population genetics in Thailand. Those studies have exclusively explored the genetic profile of the Tai people’s maternal and paternal lineages.23, 24, 25, 26, 27, 28, 29 In 2001, mitochondrial DNA (mtDNA) variations of linguistically and geographically diverse ethnic groups of Thailand, including the Kra–Dai Thai in Khon Kaen and Chiang Mai Provinces, Phutai and Black Tai, and non Kra–Dai populations (Sakai, Chong, Mussur and Lisu), indicated genetic distinction of the Sakai, whereas genetic relatedness was observed between Chong and Black Tai. The two Thai populations showed close genetic affinity to Phutai, Mussur and Lahu, indicating an absence of population grouping based on geography and language.23 Later, the maternal genetic structure of four Kra–Dai ethnicities, that is, Yuan, Yong, Khuen and Lue from Northern Thailand were investigated. The main evolutionary force, such as genetic drift, influenced genetic differentiation in these four Kra–Dai populations, possibly resulting in genetic and linguistic inconsistency.24 Lertrit et al.25 reported mtDNA variation in ancient samples and extant Austroasiatic populations (Chaobon and Khmer) in Northeastern Thailand with a genetic comparison with multiple Kra–Dai populations in Southeast Asia. Genetic links among Khmer and Kra–Dai had been observed, possibly by population admixture, whereas the Chaobon had genetically differentiated from other Kra–Dai groups.25 Kutanan et al.26, 27 evaluated mtDNA and Y-STRs diversity in various Northern Kra–Dai and Austroasiatic groups. Sex-bias admixture has been suggested as an important factor in shaping the non-identical demographic history of the Khon Meang, the largest population in Northern Thailand.26
The genetic structure of Northern Thai Austroasiatic and Kra–Dai populations have been evaluated to estimate the degree of their genetic divergence and their internal structure. The Austroasiatic populations revealed genetic divergence among each other and also when compared with neighboring Kra–Dai peoples. Correlation between genetics and language was also presented in this study.27 MtDNA variability in Northeastern Thai populations, recently scrutinized by Kutanan et al.28, was used to calculate the genetic contribution from parental Lao and Khmer to the Thai–Isan people who constitute the majority in Northeastern Thailand.28 More recently, the same group also evaluated the relative role of geographic barriers and linguistic differences as possible causes affecting the maternal genetic distances among 10 Northeastern Thai ethnicities, including the Kra–Dai (Kaleung, Phu Thai, Saek, Nyo and Lao Isan) and Austroasiatic peoples (So, Suay, Khmer, Chaobon and Mon). In their divergence of genetic differences, geography was considered as the most important factor.29
Although genetic diversity in Thai populations has been inspected by several studies, only a uniparentally inherited maker was utilized. Data from biparentally inherited genetic marker systems have been limited so far. In addition, those previous literatures pivotally investigated populations from the North and Northeast regions, although overlooking Central Thai populations. Among the 62 ethnolinguistic groups of Thailand, around 24 speak Kra–Dai languages,30 only 5 of which have been genotyped for autosomal microsatellites or short tandem repeats (STRs).31, 32 To obtain a more comprehensive knowledge of the Kra–Dai-speaking populations in Thailand, we have extended the study to 11 new Kra–Dai-speaking populations belonging to nine ethnolinguistic groups, namely the Kaleung, Phu Thai, Saek, Nyo, Lao Isan, Yuan, Black Tai, Lue and Phuan. The questions of general relevance scrutinized here are as follows: (1) how the genetic diversity and genetic relationship among these populations compare with previously researched the Kra–Dai-speaking groups, and (2) whether geographic and linguistic factors are important in determining genetic variation in the studied populations.
Materials and methods
Samples
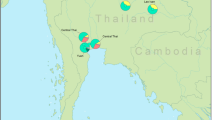
Five hundred individuals from 11 populations belonging to nine ethnicities were examined: the Kaleung (KAL, n=47), Phu Thai (PUT, n=42), Saek (SAK, n=27), Nyo (YOH, n=48), Lao Isan (LAO, n=45), Yuan from the Provinces of Uttaradit (YU-Ut, n=79) and Ratchaburi (YU-Ra, n=45), Black Tai (BTA, n=42), Phuan (PUA, n=37), and Lue from the Provinces of Chiang Rai (LU-Cr, n=47) and Chiang Mai (LU-Cm, n=41). Information on linguistic, culture and individual history from interviews was used to ensure that none of the individuals were related for at least three generations and had a non-mixed ethnicity. The studied populations reside in three regions of Thailand, Northern Thailand, Northeastern Thailand and Central Thailand (Table 1 and Figure 1).

Map of Thailand showing the location of studied populations in different regions. (Filled circles: the present populations studied, blank circles: the compared populations from earlier study.) Population codes are given in Table 1.
Informed consent was obtained from subjects before buccal swabs collection using a brush embedded in a Gentra Puregene Buccal Cell Kit (Qiagen, Hilden, Germany). Genomic DNA was extracted from the collected buccal cells using the same kit according to the manufacturer's protocols. For the Lue samples, DNA was obtained from Kampuansai et al.24 The Ethics Committee for Human Research of Khon Kaen University and Naresuan University, Thailand approved the present study’s protocol for the use of human subjects.
STR typing
One to 5-ng DNA templates were amplified for 15 autosomal STR loci as follows: D8S1179, D21S11, D7S820, CSF1PO, D3S1358, THO1, D13S317, D16S539, vWA, TPOX, D18S51, D5S818, FGA, D19S433 and D2S1338 using a commercial AmpFℓSTR Identifiler kit (Applied Biosystem, Foster City, CA, USA). PCR conditions were carried out as recommended by the manufacturer’s protocol. Amplicons were genotyped by multicapillary electrophoresis in an ABI3130 genetic analyzer (Applied Biosystem). STR alleles were then designated by GeneMapper software v.3.7 (Applied Biosystem) and compared with the allelic ladder supplied with the kit.
Statistical analyses
An ARLEQUIN software package v. 3.533 was employed to compute allele frequencies at each locus, the number of alleles, Hardy–Weinberg P-values, observed heterozygosity, expected heterozygosity (HE), gene diversity (GD) values, and the mean number of pairwise differences (MPD) within and between populations. Bonferroni correction of the P-value was applied (α=0.05/15 or 0.0033). Multiple statistics of forensic genetic relevance, for example, matching probability, power of discrimination, power of exclusion, typical paternity index and polymorphism information content, were calculated using the Excel Power-Stats spread sheet.34
Population affinity was assessed by two distance-based clustering approaches. Pairwise genetic distances based on normalized allele frequency variance (Fst) and their statistical significance using 1000 permutations were estimated by ARLEQUIN. The Fst distance matrix was then plotted in two dimensions by means of multidimensional scaling, using STATISTICA 10.0 software (StatSoft). In addition, principal coordinates analysis (PCoA) was conducted by a covariance matrix with data standardization by GENALEX 6.3 software35 and utilized to reveal genetic relationship.
To investigate the cryptic population structure, the Bayesian clustering method was implemented in STRUCTURE 2.336, 37, 38 under assumptions of admixture, correlated allele frequencies and using the LOCPRIOR model.36 For each number of clusters (K) running from 1 to 12, five replications were performed, using an MCMC chain burn-in length of 100 000 iterations followed by a 400 000 iteration run length to ensure chain convergence.
Inference for populations groups, which show geographic and genetic similarity, was performed by spatial analysis of molecular variance (SAMOVA) in a SAMOVA v.1.0 program.39
An analysis of molecular variance (AMOVA)40 using ARLEQUIN calculated genetic variance at three hierarchical subdivisions (within individuals of populations, among populations within a group and among groups of populations) according to region. The Mantel test was performed to test correlations and partial correlations between three matrices of genetic, geographic and linguistic distances. Geographic distances in the form of great-circle distances among populations were calculated from their latitudinal and longitudinal coordinates (Supplementary Material 1). For linguistic distance, except for the language of SAK, which belongs to the Northern sub-branch, all populations were linguistically grouped in the Southwestern sub-branch. Because detailed hierarchical classification of Southwestern languages is still a debatable issue,41, 42, 43, 44, 45 linguistic distances between pairs of populations were defined according to the following criteria. Populations speaking dialects of the same language, for example, LU-Cr-YO, were assigned a dLAN of one. In contrast, populations speaking languages that clearly belong to the same immediate Southwestern subgroup, for example, YOH-KAL, were given a dLAN of two. Furthermore, populations speaking Southwestern languages that could not be definitely placed within the same immediate subgroup were given a dLAN of three. Last, a pair of populations that included the non-Southwestern language SAK was assigned a dLAN of four (Supplementary Material 1).
The relative amount of gene flow into each population was assessed according to the R-matrix model of Harpending and Ward,46 which involved a regression analysis of mean per locus heterozygosity on the distance from the centroid (rii).46
To support whether language or geography can better explain the genetic variation of the Kra–Dai-speaking groups in Thailand, linguistic and geographic models were constructed (Figure 2). In the linguistic model, all studied populations were combined in a panmictic population that had a migration rate among them equal to one, as well as those populations originating in a period of time corresponding to the origin of the Tai language (T1 and T2). In contrast, the three geographically distinct populations, those from the North, the Central and the Northeast, were separated according to the historical migrations.

Demographic models for approximate Bayesian computation (ABC) analysis correspond to language and geography. Details of parameters used are shown in Supplementary Material 2.
In both models, effective population sizes were assumed to be constant in time, and that the prior distributions were all uniform (log-uniform for the effective population sizes) and where possible based on the historical records (Supplementary Material 2). The simulation-based approximate Bayesian computation (ABC) procedure was employed to select the best model. The ABC approach analyses the combination of large genetic data and realistic models. Millions of genetic data set with the same feature of the observed one, that is, number of individuals, type of genetic markers and length of the sequences, are produced according to the coalescent theory for each investigated demographic model in which model parameters can draw from the associated prior distributions. Euclidean distance was utilized to compare the pattern of genetic variation in the observed and simulated data, and then summarized by a certain number of statistics. Only the coalescent-based simulations that generated summary statistics close to the observed ones, indicated by the smallest Euclidean distances were then considered to calculate the posterior probabilities of the created demographic models.
The software package ABCtoolbox47 was employed to generate the simulated 500 000 data sets for each model. Two different approaches, the acceptance–rejection (AR) procedure48 and weighted multinomial logistic regression (LR)49 were used to compute the posterior probabilities. Under AR, the calculation of posterior probabilities considered only a certain number of ‘best’ simulations, and then simply counted the proportion of those retained simulations that were generated by each investigated model. The reliable results of this method were generated when a few simulations showing an excellent fit with the observed data was applied, for example, a few hundreds.49 Thereofore, 100, 200, 300 and 500 of the best simulations were used in this analysis.
In contrast, under the LR method, a logistic regression is fitted when the model is the categorical-dependent variable in the ABC simulations and the summary statistics are the predictive variables. The regression is local around the vector of observed summary statistics, and the probability of each model is finally evaluated at the point corresponding to the observed vector of summary statistics. The β coefficients of the regression model are estimated by maximum likelihood. The simulations were set at 75 000, 100 000, 150 000 and 200 000.The R scripts from http://code.google.com/p/popabc/source/browse/#svn%2Ftrunk%2Fscripts were modified to calculate the models’ posterior probabilities. Mean and the s.d. over loci in each population of four parameters, that is, the number of alleles, heterozygosity, modified Garza–Williamson index and the allelic range, were the utilized summary statistics.
Results
Standard diversity indices
As the set of genetic markers in this study are commonly utilized for forensic purposes, allele frequency distributions and forensic parameters of each STR locus in each population are presented in the Supplementary Materials 4 and 5. In addition, important population genetic and forensic parameters are summarized in Table 1. The statistical evaluations of the new studied populations were compared with the published population data. The average HE ranged from 0.7586 (KH) to 0.7871 (YU-Ut). The YU-Ut exhibited the highest GD (0.7871±0.3964), whereas the lowest GD was shown in PUT (0.7585±0.3850). A maximum of 133 alleles were observed in YOH and a minimum of 104 alleles in LU-N2. It is interesting that the SAK displayed a low value of HE, GD and number of alleles (0.7653, 0.7613±0.3890 and 105, respectively).
The combined matching probability ranges from 1 in 9.49 × 10−17 (YU-Sa) to 1 in 1.15 × 10−15 (KH). The combined power of exclusion is >0.9999 in all populations except the SAK (0.9979) and LU-N2 (0.9936). In each of these populations, the combined power of discrimination values were >0.99999999. Only one locus, D13S317 in LU-Cm, deviated from the Hardy–Weinberg equilibrium, with the Bonferroni correction.
Population clustering
To test for any statistically significant genetic differences in the populations examined in this study, pairwise Fst comparisons based on the number of different alleles were performed. Among 153 comparisons, 98 (64%) were statistically significant after the Bonferroni correction (P<0.00094; Supplementary Material 1). KAL and SAK showed significant Fst values for all comparisons, indicating a high genetic differentiation. The YOH, PUA, KH and LU-N2 were genetically differentiated from all other populations.
Distance-based clustering of populations revealed by the multidimensional scaling plot (Figure 3) based on the Fst distance matrix indicated that all four Yuan (YU) populations were clustered together in the center of the plot, with the LU-Cr, LU-Cm, LAO, BTA and YO, which reflects a close genetic affinity between these groups. The PUT, SH and LU-N1 were scattered intermediately between the central cloud and margins of the plot, indicating their weak genetic differentiation. The PUT and SH were closely related and both populations were segregated away from the LU-N1. Six populations (SAK, KH, LU-N2, PUA, YOH and KAL) were dispersed at the edge of the plot, reflecting a strong degree of genetic differentiation. The PCoA result was concordant with the multidimensional scaling result. SAK was an outlier at the upper end of axis 1, which explained 17.06%, whereas KH, LU-N1, LU-N2, PUA and YOH were separated from all populations on axis 2, which explained 14.62% of the variance of the distance matrix (Figure 4a). KAL, PUT, and SH were located higher up on axis 3, which explained 12.32% of the variation (Figure 4b). This result shows a degree of divergence between SAK, KH, LU-N1, LU-N2, PUA, YOH, KAL, PUT and SH from the other populations. Neither geographical nor linguistic grouping was observed in both distance-based clustering analyses.

Two-dimentional scaling plot (2D-MDS) constructed based on pairwise Fst. The stress value is 0.1759. Population codes are given in Table 1.

Principal coordinate analysis (PCoA) was performed using a covariance matrix with data standardization. Two-dimensional plot of the PCoA between axes 1 and 2 and between axes 1 and 3 were depicted in Figures 4a and b, respectively. Population codes are given in Table 1.
Model-based clustering methods were implemented in STRUCTURE 2.3 to clarify population sub-structuring. Although the analyses were executed using sampled information, which provides more information due to a low level of genetic divergence among populations (average Fst=0.0099),50 no population sub-structuring was detected. At K=2, all populations were assigned to the same cluster (cluster 1), although with a very weak degree of differentiation from PUA (membership proportion of 0.1203 in cluster 2). When K was increased to 3, the results were unchanged. Again, a new very weak cluster belonging to LU-N2 and KH emerged with a membership proportion of 0.2890 and 0.1508, respectively, in cluster 3. Although more clusters were run until K=12, no population structure was observed. Therefore, membership proportions of each population and a simulation summary for K=2 and K=3 are only represented (Table 2).
To get a picture of population clustering that is best supported by the genetic data, SAMOVA was carried out. SAMOVA results suggested the isolation of populations, which are clearly differentiated on the multidimensional scaling and PCoA plot. Using a two-groups category, the SAK was partitioned from the other populations indicating the highest degree of differentiation. When the number of groups was increased from 3 to 7 groups, the LU-N2, KAL, YOH, KH and PUA were segregated from the remaining populations (Table 3). This result indicates that LU-N2, KAL YOH, KH and PUA are the next most differentiated populations, respectively.
The AMOVA and Mantel test
The AMOVA procedure was employed to determine the proportion of total genetic variation among regions. The results indicated that population groupings based on region cannot describe the genetic structure of the studied populations, as the amount of observed variation among groups was 0.08% with no statistical significance (Fct=0.0008, P>0.01). The average Fst of Northeastern Thai populations (Fst=0.0152, P<0.01) were higher than the overall Fst (0.0099, P<0.01), whereas lower Fst values were observed in Northern and Central Thai populations (Fst=0.0008, P<0.01 and Fst=0.0060, P<0.01, respectively). AMOVA observation indicates genetic homogeneity of Northern and Central Thai populations, but reveals genetically heterogeneous Northeastern Thai populations.
On the basis of several clustering analyses, SAMOVA, and AMOVA, it seemed likely that population groupings were not correlated with region. We therefore used the Mantel test to verify our observation. A statistically insignificant correlation (r=0.0836, P>0.01) as well as a partial correlation (r=−0.0896, P>0.01) between genetic and geographic distances was detected, indicating no geographic effect on genetic diversity of the studied populations. We further quantified the effect from linguistic factors, which can drive genetic variation among populations. The Mantel test showed that among genetic and language matrices, the correlation (r=0.4532, P<0.01) and partial correlation (r=4541, P<0.01) was significant.
Model selection
The posterior probabilities from the ABC analysis of the two considered evolutionary models were the deciding factor for us to choose the linguistic model (Table 4) in agreement with previous analyses, indicating that language explains better the genetic variation in the data set. The posterior probability of the linguistic model was the highest in both AR and LR and remained stable over a different number of retained simulations. These probabilities reached a value of 100% using the LR procedure. This strongly indicates that language has had a major role in shaping the genetic variation between the Kra–Dai-speaking groups in Thailand.
R-matrix analyses
A regression plot between average heterozygosity and the distance from the centroid (rii) was constructed based on Harpending et al.46 (Figure 5), in which if populations have previously experienced from both gene flow and genetic drift in equal parts, a linear trend will be obtained given a constant effective population size and the same migration rate among those populations. The YO, BTA and LU-Cr were positioned in close vicinity of the theoretical line. All of four YU populations (YU-Ut, YU-Cm and YU-Ra), SH, YOH and PUA have received a greater than average gene flow from outside because they lie above regression line. In contrary, almost all of the Northeastern Thai (PUT, LAO, KAL and SAK) and LU populations (LU-N1, LU-N2 and LU-Cm) as well as the KH were placed below and farther from the line, suggesting that greater isolation and a less than average gene flow into them from the outside.

Plots of average heterozygosity versus distance from the centroid (rii). Dash line indicates theoretical regression line. Population codes are represented in Table 1.
Discussion
A number of molecular anthropological studies have examined contemporary Thai populations focusing on mtDNA data but have overlooked autosomal markers. This study examines the autosomal microsatellites from multiple ethnicities from three different regions of Thailand, including the North, the Northeast, and the Central, to determine the genetic affinity as well as genetic diversity, with geography and language as factors affecting human genetic relationships.
The main observation from this study is that there is genetic similarity within the Kra–Dai-speaking populations, as indicated by a low average pairwised Fst (0.0099) when compared with the various linguistic groups in the same marker, specifically, the Austroasiatic speakers in Northern Thailand (0.0244),31 and the Sino-Tibetan speakers in Southern China (0.0181),51 and in India (0.0238).52 This low degree of differentiation among the Kra–Dai-speaking groups is also substantiated by the fact that STRUCTURE did not generate any detectable population structure. Even the LOCPRIOR model, which can assist in sampling when there is a very low level of genetic distance,36 yielded no population substructure. Patterns of close genetic relationship within the Kra–Dai grouping in Thailand were also indicated by uniparentally inherited markers, like mtDNA and Y-STRs.26, 27, 31
Genetic resemblance among populations might stem from various population histories and events, for example, random genetic drift and recent gene flow originating from a common ancestral population. The haploid regions of mtDNA and non-recombining portion of the Y-chromosome (NRY) are more susceptible to random genetic drift owing to a fourfold reduction in effective population size (Ne) relative to the autosomes. Conversely, current study employs autosomal STRs, which are biparentally inherited and distributed throughout the genome, to eliminate this disadvantage. Therefore, the genetic drift should not be the main factor determining genetic relatedness in this study, except for the Saek (SAK), which will be discussed later. Furthermore, recent gene flow processes might be difficult to happen because the studied populations are located in different regions. The isolation by distance model promoting correlation between genetic variation and geographic distances is also absent, which is supported by the results from the Mantel test (r=0.0836 for correlation and r=−0.0896 for partial correlation, P>0.01 for both) and AMOVA (Fct=0.0008, P>0.01). Therefore, the interaction between genetic drift and dispersal of individuals between populations did not affect the current genetic variation patterns.
The most reasonable theory to describe our results is that the populations originated from one common ancestral population resulting in the similarity of their genetic structure. This resemblance in genes of all populations correlates to lingustic similarity, which is classified as a member of the Southwestern sub-branch within the Tai branch of the Kra–Dai language family. Again, the Saek is the only exception, as it belongs to the Northern sub-branch. The shared features of language in one group can be a result of shared genetic ancestry,53, 54, 55, 56, 57 thus, we propose that the genetic homogeneity among the studied the Kra–Dai-speaking populations suggests that they descended from the same ancestral population. If linguistic classification is a reflection of genetic history, linguistic distance possibly reveals subtle genetic differences among populations as reflected by the Mantel test. The fact that both correlation and partial correlation values between linguistic and genetic distances are very high (r=0.4532 for correlation and r=0.4541 for partial correlation, P<0.01 for both) confirms that linguistic affiliation is the primary factor in determining the genetic structure of the entire population. However, in this study, one methodological drawback is the lack of concensus on the relationship among the Southwestern Tai languages. A generally accepted and more elaborate subgrouping would allow a finer resolution of the linguistic distance to be applied in the future work. We expect the correlation between language and gene to be even stronger when this aspect of methodology is improved. To our knowledge so far, the best predictor of genetic relatedness among Kra–Dai populations was language. To confirm our assumption, the simulated results from ABC also selected demographic models in which language was tested to explain the genetic variation of the studied populations (Table 4). Agreement among results from various experiments supports the view that language is a responsible factor to determine genetic variation of populations in this study.
Although most of the Kra–Dai groups originated from a common ancestral population and migrated southward from the area of Southern or Southeastern China into the area of present-day Thailand, approximately a 1000 years ago8, 10, 58 the migratory route and reasons were different for successive waves. After a migration and settlement period, most populations still exhibited a genetically homogenous structure, but some populations developed a genetic difference from the common stock because of factors like genetic drift and perhaps admixture with other differentiated populations, as indicated by the results of the R-matrix analysis (Figure 5).
In Northern Thailand, the Yuan are regarded as the first Tai group in Thailand who settled down in 8th century CE in the area referred to as the golden triangle, the point where the borders of Laos, Thailand and Myanmar meet.59 Then, they expanded their kingdom to dominate the Northern Thai area around Chiang Mai city (YU-Cm). They constitute the majority of Northern Thailand people. Around 200 years ago, some groups of Yuan were forced to migrate southward to the lower part of Northern Thailand in the area of Uttaradit Province (YU-Ut), and some migrated even further southward to Central Thailand and settled down in the Province of Saraburi (YU-Sr) and Ratchaburi (YU-Ra).9, 60 Even though the Yuan in Uttaradit, Suraburi and Ratchaburi Province had left their historical hometown of Northern Thailand hundreds years previously, multiple results in this study indicated a similar genetic structure as the Yuan in Chiang Mai (YU-Cm).
Within the Yuan group, the Yuan from Uttaradit (YU-Ut) exhibited a high genetic diversity as reflected by the HE, GD and number of alleles, which affects the gene flow process in this population. In addition, the highest MPD within the population (Supplementary Material 3) reflected genetic heterogeneity within the population, as well as the most farthest outlier above the line as depicted in Figure 5 evidently supported the hypothesis of gene flow into YU-Ut. On the basis of the ethnolinguistic landscape, Uttaradit Province is the buffer zone between the Northern Thai language and the Central Thai language. Therefore, on the basis of the genetic and linguistic correlation, population admixture could be the relevant factor enhancing the level of genetic diversity in the YU-Ut. However, genetic variation values in the other YU populations from Central Thailand (YU-Sr and YU-Ra) were not as high as YU-Ut. The YU population in Central Thailand lived segregated from the Thai majority, resulting in less gene flow in both directions. This may explain the genetic similarity of YU-Sr to the YU-Cm despite 200 years of separation.
The Yong (YO) is another population who has a homogenous gene pool with the Yuan. The Yong left their historical hometown in Yong, the Shan State of Myanmar, as captives of war around 1805 AD24, 31 They settled in Lamphun province of Northern Thailand, which at the time was primarily occupied by the Yuan.11 Consequently, the genetic similarity of Yong and Yuan may be the result of a recent gene flow between the neighboring populations originating from a common ancestral population.
The studied Phuan population is located in Sukhothai Province in the Northern region of Thailand. The PUA migrated from Muang Phuan, XiengKhouang in Laos, an area with a long and complex history of foreign outside interference. They had to accept the suzerainty of Siam, Laos and Vietnam at the same time. As a consequence of a depopulation policy adopted by the Siamese court, the relocation of PUA people began during the late 18th and early 19th centuries.15, 16 The weak differentiation of the Phuan from other groups might be the result of external gene flow from other native Austroasiatic groups and reproductive isolation from other Kra–Dai populations, reflected by intermediate high heterozygosity and a relatively high of rii value (Figure 5).
Although the four Lue villages from Chiang Rai (LU-Cr), Chiang Mai (LU-Cm) and Nan Province (LU-N1 and LU-N2) exhibited a certain degree of heterogeneity within ethnicity, particularly the LU from Nan province that was possibly caused by founder effects as discussed in previous literature24, 26, 27, 31 and also approved by a current study in R-matrix analysis (Figure 5), the two currently studied Lue (LU-Cr and LU-Cm) were still genetically related to the Yuan. The Khuen and Shan are the other two Northern most populations and with them a weak degree of differentiation was detected. Concordant with linguistic evidence, the Khuen are genetically related to the Lue,24 whereas the Shan (SH) or great Tai, who predominantly reside in the Shan State of Myanmar and migrated into the mountainous areas of Northern Thailand around 1400 AD, have a close genetic relatedness to the Northeastern populations, especially the populations residing in the Sakon Nakorn Basin of Northeastern Thailand, that is, the Phu Thai (PT), Nyo (YOH) and Kaleung (KL). It is interesting that despite the language difference between the Shan and other Kra–Dai populations from Northeastern Thailand41 our study indicated genetic similarity. Perhaps the Shan and Kra–Dai populations from Northeastern Thailand maintain the original genes of Southwestern Tai. In contrast, the genetic admixture of these populations with the same native Austroasiatic sources might be another reason to explain the genetic relationship among them. The fact that Shan is positioned above the theoretical in the regression plot (Figure 5) suggests that this genetic source is a population that is not included in this study.
In the Northeast of Thailand, we reported five contemporary Kra–Dai populations. The majority of the inhabitants, the Lao, are widely distributed in all the provinces in Northeastern Thailand and Laos. They were forcibly removed from their historical homeland in present-day Laos during the 19th century.61 Our results indicate a close genetic affinity between the Lao and Yuan, reflecting a common ancestral population. Interestingly, the tremendous genetic divergence of the Saek (SAK), as reported by Kutanan et al.,29 can also be seen in the present study even though different genetic markers were applied. The SAK displayed low genetic diversity values (Table 1) indicating the loss of diversity possibly by genetic drift, which is likely a basis of the greatest genetic divergence (Table 3 and Figures 3 and 4). The position of SAK, which is far removed from the centroid, suggests a relatively greater effect of random genetic drift on this genetically and linguistically distinctive population (Figure 5). The genetic drift associated with the founder effect might have occurred within the last 200 years during the migration and resettlement period of the SAK, who originated in Vietnam and then moved westward to Laos and finally marched across Mekong river into Northeastern Thailand.21, 22
In Central Thailand, in addition to the forced resettlement of Yuan populations, the Black Tai or Lao Song (BTA) were also relocated to Phetburi Province in the 19th century. The historical hometown of the Black Tai is located in the area West of the Black river in Mueang Thaeng near the border of China’s Kwangsi province and the city of Dien Bien Phu, Vietnam.17 There is a very close genetic relationship between BTA and YU, as well as LAO, which points to a common genetic ancestry shared by these populations.
Although earlier studies reported genetic variation of Kra–Dai-speaking populations throughout Thailand, only the maternal lineage was examined. Our present study of bi-parental lineage provides evidence for the genetic similarity of Kra–Dai. This is possibly the result of having a common ancestral population. The best predictor of genetic relatedness among the populations was language, although the criteria for classifying subgroups of Southwestern Tai languages needs more study. The linguistic affinity in one group can be shaped by shared common ancestors. In contrast, geographic factor, which have recently been reported as the predominant factor to determine genetic variation in Northeastern Thai samples, failed to correlate with the genetic results in this study. This is likely because there is a difference in the populations studied. The other driving factors for genetic diversity in certain populations were genetic drift, which was very strong in the Saek, and gene flow, which are mirrored in the Yuan from Uttaradit Province. These evolutionary factors might have occurred during or after resettlement periods. In the future, a comprehensive study involving other linguistically distant populations such as the native Mon and Khmer is necessary to complete the picture of genetic affinity of different ethnicities in Thailand.
References
Li, F. K. in Culture in History: Essays in Honor of Paul Radin (ed Diamond S) 951–959 (Columbia University Press, New York, NY, USA, 1960).
Li, F. K. Handbook of Comparative Tai, (University of Hawai'i Press, Honolulu, HI, USA, 1977).
Pittayaporn, P. Proto-Southwestern-Tai revised: a new reconstruction. J. Southe. Asian linguistic Soc 2, 121–144 (2009).
United Nations, Department of Economic and Social Affairs, Population Division. World Population Prospects: The 2012 Revision, Volume II, Demographic Profiles (ST/ESA/SER. A/345) (2003) http://esa.un.org/wpp/documentation/publications.htm Accessed 10 September 2014.
Smalley, W. A. Linguistic Diversity and National Unity: Language Ecology in Thailand, (University of Chicago Press, Chicago, IL, USA, 1994).
Ongsakun, S. Prawattisat Lanna (Lanna history) 2nd edn (Amarin, Bangkok, Thailand, 1996).
Saraya, D. (Sri) Dvaravati: the Initial Phase of Siam’s History, (Muang Boran Publishing House, Bangkok, Thailand, 1999).
Wyatt, D. K. Thailand: a Short History, (Silkworm Books, Bangkok, Thailand, 1984).
Penth, H. A Brief History of Lanna: Civilizations of North Thailand, (Silkworm Books, Chiang Mai, Thailand, 2000).
Pittayaporn, P. Layers of Chinese Loanwords in Proto-Southwestern Tai as evidence for the dating of the spread of Southwestern Tai. Manusya. J. Humanities 20, 47–68 (2014).
Malasam, S. Khon Yong Yai Phaen Din (The Yong’s exodus), (Thammasat University Press, Bangkok, Thailand, 1997).
Sae-Ngove, S. & Mesathan, W. Thai Lue. Nakhon Pathom, Thailand, Encyclopedia of ethnic groups in Thailand, (Institute of Language and Culture for Rural Development, Mahidol University, 1998).
Kaise, R. Tai Yai Migration in the Thai-Burma border area: the Settlement and Assimilation Process 1962-1997, (M.A. thesis, Chulalongkorn University, 1999).
Duangthima, W. The Conservation of Tai-Khuen Community in Ban San Kangpla, Chiangmai Province, (M.A. thesis, Chulalongkorn University, 2005).
Eam-eium, C. A Phonological Description of Phuan at Tambon Hat Siaw, Si Satchanalai District, Sukhothai Province, (M.A. thesis, Mahidol University, 1986).
Tanprasert, P. A Language Classification of Phuan in Thailand: A Study of the Tonal System, (Ph.D. thesis, Mahidol University, 2003).
Pitiphat, S. The religion and beliefs or the Black Tai, and a note on the study of cultural origins. J.Siam Soc. 68, 29–38 (1980).
Damrongsakul, S. Thai Lue Nakhon Pathom, Thailand: Institute of Language and Culture for Rural Development. Encyclopedia of ethnic groups in Thailand, (Institute of Language and Culture for Rural Development, Mahidol University, 2003).
Koowatthanasiri, K. The Tones of Nyo,, (M.A. thesis, Chulalongkorn University, 1981).
Warangrat, S., Kunnawong, T. & Inthisaeng, S. Kaloeng Ban Bua: rayngan kansueksawichai Sakonnakhon, Thailand, (Research report, Arts and Cultural Center, Sakonnakhon Teacher’s College, 1988).
Chamberlain, J. R. The Origin of the Seak Implications For Tai and Vietnamese History (ed Burusphat S) 97–127 Proceedings of the International Conference on Tai Studies, 29–31 July 1998 (Institute of Language and Culture for Rural Development, Mahidol University, Bangkok, 2000).
Gedney, W. J. The Seak language of Nakhon Phanom Province. J. Siam Soc. 58, 67–87 (1970).
Fucharoen, G., Fucharoen, S. & Horai, S. Mitochondrial DNA polymorphisms in Thailand. J. Hum. Genet. 46, 115–125 (2001).
Kampuansai, J., Bertorelle, G., Castri, L., Nakbunlung, S., Seielstade, M. & Kangwanpong, D. Mitochondrial DNA variation of Tai speaking peoples in Northern Thailand. ScienceAsia 33, 443–448 (2007).
Lertrit, P., Poolsuwan, S., Thosarat, R., Sanpachudayan, T., Boonyarit, H. & Chinpaisal, C. et al. Genetic History of Southeast Asian Populations as Revealed by Ancient and Modern Human Mitochondrial DNA Analysis. Am. J. Phys. Anthropol. 137, 425–440 (2008).
Kutanan, W., Kampuansai, J., Nakbunlung, S., Lertvicha, P., Seielstad, M. & Bertorelle, G. et al. Genetic structure of Khon Mueang populations along a historical Yuan migration route in Northern Thailand. Chiang Mai J. Sci. 38, 295–305 (2011a).
Kutanan, W., Kampuansai, J., Fuselli, S., Nakbunlung, S., Seielstad, M. & Bertorelle, G. et al. Genetic structure of the Mon-Khmer speaking groups and their affinity to the neighbouring Tai populations in Northern Thailand. BMC Genet. 12, 56 (2011).
Kutanan, W., Srithawong, S., Kamlao, A. & Kampuansai, J. Mitochondrial DNA- HVR1 variation reveals genetic heterogeneity in Thai-Isan peoples from the lower region of Northeastern Thailand. Adv. Anthropol. 4, 7–12 (2014).
Kutanan, W., Ghirotto, S., Bertorelle, G., Srithawong, S., Srithongdaeng, K. & Pontham, N. et al. Geography has more influence than language on maternal genetic structure of various northeastern Thai ethnicities. J. Hum. Genet. 59, 512–520 (2014).
Premsrirat, S., Deepadung, S., Buasuang, A., Suwanket, E., Choosri, I. & Srijampa, S. et al Ethnolinguistic maps of Nakhon Pathom, Thailand, (Institute of Language and Culture for Rural Development, Mahidol University, 2004).
Kutanan, W., Kampuansai, J., Colonna, V., Nakbunlung, S., Lertvicha, P. & Seielstad, M. et al. Genetic affinity and admixture of northern Thai people along their migration route in northern Thailand: evidence from autosomal STR loci. J. Hum. Genet. 6, 130–137 (2011).
Kutanan, W. & Kampuansai, J. Genetic variation of the Yuan in Saraburi province of central Thailand revealed by autosomal forensic STRs. Chiang Mai J. Sci. 41, 39–47 (2014).
Excoffier, L. & Lischer, L. Arlequin suite ver 3.5: A new series of programs to Perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Excel PowerStats spread sheet www.promega.com/geneticidtools/powerstats. Accessed 1 July 2014.
Peakall, R. O. D. & Smouse, P. E. Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 88–295 (2006).
Hubisz, M., Falush, D., Stephens, M. & Pritchard, J. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332 (2009).
Pritchard, J.K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Falush, D., Stepheas, M. & Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164, 156–1587 (2003).
Dupanloup, I., Schneider, S. & Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 11, 2571–2581 (2002).
Excoffier, L., Smouse, P. & Wuattro, J. Analysis of molecular variance inferred from metric distance among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491 (1992).
Pittayaporn, P. Tonal developments and Southwestern Tai subgrouping. J. Lett. 42, 305–339 (2013).
Brown, M.J. in From Ancient Thai to modern dialects, and other writings on historical Thai linguistics (ed Brown M. J.) 69–254 (White Lotus, Bangkok, Thailand, 1965).
Chamberlain, J.R. in Studies in Tai Linguistics in Honor of William J. Gedney (eds Harris J. G. & Chamberlain J. R.) 49–66 (Central Institute of English Language, Bangkok, Thailand, 1975).
Jonsson, N. L. Proto-Southwestern Tai, (Unpublished Doctoral dissertation, Albany, NY, State University of New York at Albany, 1991).
Kullavanijaya, P. & L-Thongkum, T. Linguistic Criteria for Determining Tai Ethnic Groups: Case Studies on Central and South-Western Tais (eds Burusphat S) 273–298 Proceedings of the International Conference on Tai Studies, 29–31 July 1998 (Institute of Language and Culture for Rural Development, Mahidol University, 2000).
Harpending, H. C. & Ward, R. in Biochemical Aspects of Evolutionary Biology (ed Nitecki M) 213–256 (University of Chicago Press, Chicago, 1982).
Wegmann, D., Leuenberger, C., Neuenschwander, S. & Excoffier, L. ABCtoolbox: a versatile toolkit forapproximate Bayesian computations. BMC Bioinformatics 11, 116 (2010).
Pritchard, J. K., Seielstad, M. T., Perez-Lezaun, A. & Feldman, M. W. Population growth of human Y chromosomes: a study of Y chromosome microsatellites. Mol. Biol. Evol. 16, 1791–1798 (1999).
Beaumont, M. in Simulation, Genetics and Human Prehistory (eds Matsumura S., Forster P. & Renfrew C) 135–154 (McDonald Institute for Archaeological Research, Cambridge, England, 2008).
Latch, E. K., Dharmarajan, G., Glaubitz, J. C. & Rhodes, O. E. Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation conservation. Genetics 7, 295–302 (2006).
Kraaijenbrink, T., Zuniga, S., Su, B., Shi, H., Xiao, C. J. & Tang, W. R. et al. Allele frequency distribution of 21 forensic autosomal STRs in 7 populations from Yunnan, China. Forensic Sci. Int. Genet. 3, e11–e12 (2008).
Krithika, S., Maji, S. & Vasulu, T. S. A Microsatellite Guided Insight into the Genetic Status of Adi, an Isolated Hunting-Gathering Tribe of Northeast India. PLoS ONE 3, e2549 (2008).
Barbujani, G. & Sokal, R. Zones of sharp genetic change in Europe are also linguistic boundaries. Proc. Natl Acad. Sci. USA 87, 1816–1819 (1990).
Barbujani, G. DNA variation and language affinities. Am. J. Hum. Genet. 61, 1011–1014 (1997).
Soka, R. R. Genetic, geographic, and linguistic distances in Europe. Proc. Natl Acad. Sci. USA 85, 1722–1726 (1997).
Cavalli-Sforza, L. L. Genes, Peoples and Languages. Proc. Natl Acad. Sci. USA 94, 7719–7724 (1997).
Nettle, D. & Harris, L. Genetic and linguistic affinities between human populations in Eurasia and West Africa. Hum. Biol. 75, 331–344 (2003).
O’Connor, R. Agricultural change and ethnic succession in Southeast Asian states: a case for regional anthropology. J. Asian. Stud. 54, 968–996 (1995).
Ongsakul, S. History of Lan Na, (Silkworm Books, Chiang Mai, Thailand, 2005).
Muenjanchoey, R. Thai Yuan. Nakhon Pathom, Thailand, Encyclopedia of ethnic groups in Thailand, (Institute of Language and Culture for Rural Development, Mahidol University, 2005).
Schliesinger, J. Tai Group of Thailand, Volume 1: Introduction and Overview, (White Lotus Press, Bangkok, Thailand, 2001).
Acknowledgements
We are deeply grateful to all volunteers for contributing buccal swabs samples. We thank Dr Jatupol Kampuansai for donating the Lue’s DNA samples. SS was supported by Faculty of Science, Khon Kaen University. MS was supported by four financial groups as follows: (1) Naresuan University, (2) Department of Molecular and Medical Genetics, University of North Texas Health Science Center at Fort Worth, Texas, (3) the Higher Education Research Promotion and National Research University, Office of the Higher Education Commission and (4) Thailand Research Fund (Grant number MRG5480012). PP was supported by the Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University (RES560530179-HS). SG was supported by the European Research Council, ERC Advanced Grant Agreement No. 295733, ‘LanGeLin’ Project. WK was funded by the Institute for the Promotion of Teaching Science and Technology (IPST) under the Research Fund for DPST Graduates with First Placement (Grant number 039/2555).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Srithawong, S., Srikummool, M., Pittayaporn, P. et al. Genetic and linguistic correlation of the Kra–Dai-speaking groups in Thailand. J Hum Genet 60, 371–380 (2015). https://doi.org/10.1038/jhg.2015.32
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.32
This article is cited by
-
Forensic and genetic characterizations of diverse southern Thai populations based on 15 autosomal STRs
Scientific Reports (2022)
-
Close genetic relationship between central Thai and Mon people in Thailand revealed by autosomal microsatellites
International Journal of Legal Medicine (2021)
-
Complete mitochondrial genomes of Thai and Lao populations indicate an ancient origin of Austroasiatic groups and demic diffusion in the spread of Tai–Kadai languages
Human Genetics (2017)
