
Abstract
Silver–Russell syndrome (SRS) is a heterogeneous disorder characterized by intrauterine and post-natal growth retardation, dysmorphic facial features and body asymmetry. About 50% of the patients carry (epi)genetic alterations involving chromosomes 7 or 11.The high proportion of patients with unidentified molecular etiology suggests the involvement of other genes. Interestingly, SRS patients share clinical features with the 12q14 microdeletion syndrome, characterized by several deletions with a 2.6 Mb region of overlap. Among the genes present in this interval, high mobility AT-hook 2 (HMGA2) appears to be the most likely cause of the growth deficiency, due to its described growth control function. To define the role of HMGA2 in SRS, we looked for 12q14 chromosome imbalances and HMGA2 mutations in a cohort of 45 patients with growth retardation and SRS-like phenotype but no 11p15 (epi)mutations or maternal uniparental disomy of chromosome 7 (matUPD7). We identified a novel 7 bp intronic deletion in HMGA2 present in heterozygosity in the proband and her mother both displaying the typical features of SRS. We demonstrated that the deletion affected normal splicing, indicating that it is a likely cause of HMGA2 deficiency. This study provides the first evidence that a loss-of-function mutation of HMGA2 can be associated with a familial form of SRS. We suggest that HMGA2 mutations leading to haploinsufficiency should be investigated in the SRS patients negative for the typical 11p15 (epi)mutations and matUPD7.
Similar content being viewed by others


Introduction
The Silver–Russell syndrome (SRS, OMIM180860) is a clinically and genetically heterogeneous disorder characterized by severe intrauterine growth retardation and post-natal growth retardation (PNGR), relative macrocephaly, skeletal asymmetry, typical facial dimorphisms including prominent forehead and triangular face, and additional features. The clinical spectrum of SRS ranges from severely affected patients to individuals with very mild features and although a clinical scoring system to assist the diagnosis has recently been suggested1 the accuracy of diagnosis is influenced by the experience of the clinical investigator. Autosomal dominant, autosomal recessive and X-linked inheritance models of SRS have been reported.2 Given the complex genetic etiology, it is expected that a number of different loci are implicated as cause of SRS. Recent findings have shown that the cluster of imprinted genes on chromosome 11p15.5 have a major role in this disorder. Up to 50% of SRS patients show hypomethylation of the imprinting control region 1, resulting in decreased expression of IGF2 (encoding a potent fetal growth factor) and increased expression of H19 (coding for an untranslated RNA with growth retardation property). Another 10% of the patients carry maternal uniparental disomy of chromosome 7 (matUPD7).2 A few cases with chromosomal rearrangements have been reported. At least 40% of the clinically diagnosed SRS patients have still unknown genetic etiology, suggesting the involvement of other genes.
Interestingly, SRS shares clinical features with the recently defined 12q14 microdeletion syndrome. The first evidence was reported by Menten et al.:3 this syndrome is characterized by failure to thrive in infancy, short stature, osteopoikilosis and mental retardation. The three reported unrelated patients carried deletions ranging from 3.44 to 6 Mb, with a 3.44 Mb common deleted region. The deleted interval contains 13 Ref Seq genes, including LEMD3, which is the causal gene for osteopoikilosis, glutamate receptor interacting protein 1 (GRIP1) involved in glutameric synaptic transmission and high mobility AT-hook 2 (HMGA2) with an important role in growth regulation. In 2009, Buysse et al.4 reported two additional individuals with the microdeletion syndrome and a family with proportionate short stature cosegregating with a ~170 kb deletion including the HMGA2 gene. In 2010, Spengler et al.5 identified a patient with a 12q14 microdeletion in a cohort of 20 SRS patients. Lynch et al.6 described two cases with a 10 Mb deletion encompassing the genes LEMD3, HMGA2, IRAK3 and GRIP1. Both of these girls demonstrated developmental delay, failure to thrive at birth, severe growth restriction and speech delay or complete lack of speech. Finally, Takenouchi et al.7 and Alyaqoub et al.8 described two patients with SRS-like phenotype and 4 Mb deletions including HMGA2.
To investigate the possible role of HMGA2 in the etiology of SRS, we screened a cohort of 45 patients with growth retardation and SRS-like phenotype but no 11p15 (epi)mutations or matUPD7 for the presence of 12q14 imbalances and HMGA2 mutations. We found a familial case in which a 7 bp deletion of HMGA2 intron 4, eliminating the 3′ AG-splicing site, cosegregated with the SRS phenotype. The microdeletion was absent in the mutation databases and was not detected in 50 healthy controls. A minigene assay was allowed to demonstrate that the mutation affected the splicing. This is the first point mutation causing loss-of-function of HMGA2 reported so far. The results further consolidate the hypothesis that haploinsufficiency of HMGA2 causes growth retardation and indicate a novel molecular etiology for familial SRS.
Materials and Methods
Patient cohort
For the present study,45 patients with pre/post-natal growth retardation and absence of (epi)mutation at 11p15 or matUPD7 were selected. About 27 of them met the clinical criteria of SRS for the presence of intrauterine growth retardation (birth weight or length ⩽−2 SDS for gestational age) and/or post-natal growth retardation (height below or equal to −2 SDS) and at least two of the following clinical signs: relative macrocephaly at birth, classical facial features and skeletal asymmetry; 9 patients with one of these signs in addition to the presence of post-natal growth retardation were considered affected by a SRS-like phenotype;9, 10 9 patients were classified as small for gestational age for the presence of isolated intrauterine growth retardation. Informed consent was obtained from all the individuals and/or their relatives. Genomic DNA of the patients and their relatives was isolated from peripheral blood lymphocytes by using the commonly used salting out procedure.
Analysis of copy number variants
Single nucleotide polymorphism (SNP) array analysis was performed by using the Gene Chip Human Mapping 250 K NspI and Cytogenetics Whole-Genome 2.7 M Arrays (Affymetrix, SantaClara, CA, USA). Labeling, hybridization, washing, scanning and image extraction were performed in accordance with the manufacturer’s protocol, while SNP copy number was assessed by using the software Genotyping Console Software 4.0 and Chromosome Analysis Suite 2.0 (Affymetrix) by a standard Hidden Markov Model method. The presence of copy number variants at chromosome 12q14.3-q15 was further tested by typing for the microsatellite markers, D12S1702, D12S868, D12S1601, D12S1294 and D12S1686. Microsatellites were amplified and the PCR products were separated by electrophoresis on a denaturing 8 M urea-8% polyacrylamide gel. A segregation analysis was carried out in those cases for which the DNA from parents was available. Information on primer sequences and marker locations were obtained from the Ensembl Genome Browser (www.ensembl.org) and the NCBI—UniSTS database (http://www.ncbi.nlm.nih.gov/unists).
HMGA2 mutation analysis
In the patients and 50 normal controls, the coding exons and exon–intron boundaries of HMGA2 (NC_00012) were amplified by PCR and sequenced. Primers and PCR conditions used for the analysis are described in Table 1. Sequencing of the PCR products was performed by PRIMM (Milan, Italy).
Splicing assay
To test the effect of the 7 bp deletion on splicing efficiency, the wild-type and mutant sequences were cloned into the RHCglo Minigene Plasmid and analyzed as described by Singh and Cooper.11 Three different mutant constructs (ΔA, ΔB and ΔC) covering different lengths of the intron IV–exon V junction were generated by inserting three synthetic double stranded oligonucleotides (PRIMM) between the SalI and BamHI sites and replacing the 3′ acceptor site of the first intron of the RHCglo plasmid. The oligonucleotides were 45-bp (wt, hg19: chr12: 66356980-66357024), 38-bp (ΔA, same sequence as wt but missing the 7 bp at the 3′ end), 41-bp (ΔB, same sequence of ΔA plus first 3 bp of exon 5) and 53-bp (ΔC, same sequence as ΔA plus first 15 bp of exon 5) long, respectively. Their sequences are reported in Table 2. The correctness of the wild-type and mutant constructs was verified by DNA sequencing (PRIMM). The constructs were individually transfected into NIH3T3 cells. Cells were cultured in Dulbecco’s modified Eagle’s medium (Sigma, St Louis, MO, USA) supplemented with 10% of fetal bovine serum, 50 U of penicillin and 50 μg ml−1 of streptomycin at 37 °C under an atmosphere of 5% CO2. The day before transfection, cells were seeded in 6-well plates at a density of 1 × 105 cells per well. About 4 μg of plasmid DNA in 250 μl Opti-MEM (Invitrogen-Life Technologies Italia, Monza, Italy) was added to an equal volume of the same medium containing 10 μl Lipofect AMINE 2000 reagent (Invitrogen-Life Technologies). The mixture was incubated for 20 min at room temperature and added to each dish. About 48 h following transfection, cells were washed with PBS, scraped and harvested by centrifugation. Three independent transfections were carried out for each construct. Cell pellets were lysed in Trizol (Invitrogen-Life Technologies) and total RNA was extracted according to the standard protocol. One microgram of total RNA was treated with RNase-free DNase (Promega Italia, Milan, Italy), and first-strand complementary DNA was synthesized using the Superscript II Reverse Transcriptase (Invitrogen-Life Technologies) and random hexamers as primers, according to the protocol of the manufacturer. Complementary DNA was amplified by using the RSV5U upstream primer (5′-CATTCACCACATTGGTGTGC-3′) and the TNIE4 downstream primer (5′-AGGTGCTGCCGCCGGGCGGTGGCTG-3′) located in vector exon 1 and exon 3, respectively, as described by the Singh and Cooper.11 Amplification products were resolved on 3% Metaphor Agarose (Lonza, Rockland, ME, USA) and stained with ethidium bromide.
Results
To look for imbalances of chromosome 12q14.3-q15, we analyzed a cohort of 45 patients with syndromic or isolated growth retardation (27 SRS, 9 SRS-like and 9 small for gestational age) and without known genetic/epigenetic alterations at chromosome 7 and 11p15 by CGH-SNP array and microsatellite markers typing (D12S1702, D12S868, D12S1601, D12S1294 and D12S1686). However, only non-pathogenic copy number variants were identified (data not shown). The same cohort was then screened for the presence of point mutations in the HMGA2 gene. For this purpose, the coding exons and the exon–intron boundaries were PCR amplified and sequenced. A deletion of 7 bp was identified in a patient belonging to a three generation-family with the clinical features of SRS (Figure 1a). Cloning and sequencing of the amplified mutant allele was allowed to determine that the microdeletion eliminated the 3′ AG acceptor site of the HMGA2 intron 4 (Figure 1b). The mutation was observed in heterozygosity in the proband and her affected mother. The maternal aunt and grandfather were also affected but unavailable to molecular analysis. Genetic material from the non affected father, maternal uncle and grandmother was also not available. The 7-bp deletion was not reported in the mutation databases (https://decipher.sanger.ac.uk/; http://www.hgmd.cf.ac.uk/ac/index.php) and was not identified by DNA sequencing in 50 healthy individuals from the Italian population.

Pedigree of the family with the 7-bp deletion and details of the mutation. (a) Three-generation pedigree of the SRS family showing co-segregation of the HMGA2 deletion with the SRS phenotype in II-3 and III-1 individuals. Individuals unavailable to molecular analysis are indicated with an asterisk. (b) The nucleotide sequences of intron 4 and exon 5 boundary from the wild-type (wt) and mutant (Δ7 bp) alleles are represented (intron sequence in lower case and exon sequence in upper case) within the structure of the HMGA2 gene. In the lower part of the figure, three electropherograms show the sequences of the cloned wild-type and mutated alleles and the genomic sequence of the proband. HMGA2, high mobility AT-hook 2; SRS, Silver–Russell syndrome. A full color version of this figure is available at the Journal of Human Genetics journal online.
The point mutation in CDKN1C gene (c.836G>[G;T]) associated with a family history of SRS as described by Brioude et al.12 was excluded by amplifying and sequencing the exons of the gene in both the proposita and her mother.
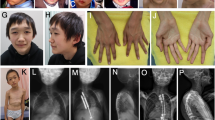
The proposita was a girl born to unrelated parents at 35 weeks of gestation. Intra-uterine growth retardation and relative macrocephaly at birth were observed (weight= 1350 g (SDS= −2.5), height= 42.5 cm (SDS= −1.3) and head circumference= 31 cm (SDS= −1)). Physical examination at 20 months (Figures 2a and b), revealed post-natal growth retardation, characteristic face with triangular form and broad and prominent forehead, thin upper lip, micrognathia, brachy-clinodactyly V of the hands, syndactyly II–III of the feet, feeding difficulties during early childhood, gastro-esophageal reflux, muscular hypotrophia, cafè au lait spot, epicanthus, clitoral hypertrophy and hypertelorism were observed. Growth retardation was also observed at age of 4 years (Figures 2c and d). Overall, a score of 12 points reflecting a severe phenotype were calculated according to the scoring system described by Bartholdi et al.10 for the SRS phenotype. Her mother also presented low stature (−3.87 SDS), triangular face with prominent forehead and brachydactyly V, and referred to have suffered of post-natal growth retardation and feeding difficulties during early childhood. Growth retardation was also referred in the maternal aunt and maternal grandfather.

Silver–Russell phenotype in the proposita at 20 months (a and b) and 4 years of age (c and d). Note the triangular shape of the face with broad forehead, small chin, thin-lipped mouth and a very thin body. Growth parameters at 20 months: weight= 6850 g (SDS=−5.7), height=72.5 cm (SDS=−3.2), head circumference= 44.2 (SDS=−2.2), BMI= 12.9 (SDS=−2.8). Growth parameters at 4 years: weight= 10800 g (SDS=−3.8), height= 88 cm (SDS=−2.9), head circumference= 46 (SDS=−2.2), BMI= 13.9 (SDS=−1.5). BMI, body mass index. A full color version of this figure is available at the Journal of Human Genetics journal online.
Since the microdeletion leaves intact two exonic AG dinucleotides close to the breakpoint that could serve as alternative acceptor site, we decided to assess the effect of the mutation on HMGA2 splicing. Since RNA was unavailable, we tested the mutant sequence by using a splicing assay in transfected cultured cells.11 Three fragments derived from the mutant allele and including the DNA sequences from position −45 of intron IV to the deletion breakpoint (ΔA), from position −45 to +3 (ΔB) and from position −45 to +15 (ΔC), respectively, as well as the wild-type sequence from position −45 to −1 were inserted into the vector to replace the endogenous 3′ acceptor site (Figure 3a). The ΔB and ΔC constructs were generated to test the possibility that the two AG dinucleotides could serve as alternative acceptor sites. The four constructs were transfected into NIH3T3 cells and splicing was analyzed by reverse transcriptase–PCR and gel electrophoresis. The results are shown in Figure 3. Two bands were observed in the lane of the empty vector RHCglo: the 111 fragment and the 79 bp fragment corresponding to the alternative spliced variants (a) and (b), respectively. The (a) variant derives from the splicing of exons 1, 2 and 3. The (b) variant derives from the alternative splicing of exon 1 with exon 3 (Figure 3a). The (a) variant was the only band observed in the lane of the wt construct, indicating that the acceptor sequence of the wt HMGA2 intron IV is correctly recognized. However, fragment (a) was absent in the lanes corresponding to the ΔA, ΔB and ΔC constructs, and only band (b) or fragments of higher molecular weight were evident, indicating that the 7 bp deletion severely affects the splicing efficiency of HMGA2 (Figure 3b).

Minigene splicing assay. (a) Structure of the minigene into the RHCglo plasmid and sequences of the HMGA2 gene analyzed in this study. The minigene exons are shown as orange boxes. The two alternative splice variants are also shown: (a) derived from the removal of intron I and II and the junction of exon 1, 2 and 3; (b) derived from the removal of intron I, exon 2 and intron II, and the junction of exon 1 with exon 3. The primers used for RT–PCR are indicated by arrows. The wild-type (wt) and mutant (ΔA, ΔB and ΔC) sequences were inserted between the SalI and BamHI sites. The wt insert includes the last 45 nucleotides of the HMGA2 intron 4, the ΔA insert includes the same sequence of wt without the last 7 nucleotides, the ΔB insert includes the same sequence of ΔA plus the first 3 nucleotides of exon 5 and the ΔC insert includes the same sequence of ΔA plus the first 15 nucleotides of exon 5. The acceptor dinucleotides AG of intron IV are in green, while the putative alternative acceptor dinucleotides of exon 5 are in blue and underlined. (b) Gel electrophoresis of RT–PCR products obtained from NIH3T3 cells transfected with the minigene constructs. The 111 bp fragment corresponding to the splice variant (a); The 79 bp band corresponds to the alternative splice variant (b). Fragments with lower mobility correspond to incorrectly spliced transcripts. Cells transfected with the original RHCglo plasmid and untransfected cells (NT) were analyzed as controls. Note that the correctly spliced transcript is generated by the wt but not by the mutant constructs. RT–PCR, reverse transcription PCR. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
In the present study, we have looked for copy number variants and point mutations of the HMGA2 gene in a group of patients affected by syndromic and non-syndromic pre/post-natal growth retardation. This analysis has led us to identify a new 7 bp intronic deletion in the HMGA2 gene in two individuals of a family with the clinical characteristics of SRS. The mutation was absent in 50 normal controls and not reported in the mutation databases. By using a minigene system and transient transfection experiments, we further demonstrated that the mutation affected the splicing and could therefore lead to haploinsufficiency of HMGA2.
HMGA2 is a transcription factor involved in a wide variety of cellular processes and with a clear role in somatic growth control. In the mouse, a ‘null’ mutation in Hmga2 is responsible for the semidominant 'pygmy' phenotype.13 In the human, chromosome aberrations leading to gain-of-function of HMGA2 are associated with overgrowth and mesenchymal tumors,14 while a common SNP in HMGA2 is associated with height variation in the general population.15 Furthermore, a 20 bp deletion in the HMGA2 intron 3 has been found in a patient with isolated growth hormome deficiency,16 and haploinsufficiency of HMGA2 is believed to be responsible for the growth retardation of the 12q14 microdeletion syndrome.17 The present study provides the first evidence that a loss-of-function mutation of HMGA2 can also be associated with a familial case with SRS phenotype. In Table 3, the clinical features associated with 12q14 microdeletion are compared with those of 11p15 ICR1 hypomethylation and matUPD7 cases. If we exclude the cases with osteopoikilosis that are associated with deletions including LEMD3, it is evident that the phenotype of the smaller 12q14 microdeletion cases can be confused with that of common SRS etiology. More cases are needed to establish if more subtle differences exist between the different molecular classes.
We suggest that HMGA2 mutations leading to haploinsufficiency might be a rare cause of SRS and should be investigated in the SRS patients that are found negative for the typical 11p15 (epi)mutations and matUPD7. In addition, HMGA2 mutations should be investigated in cases with family history of SRS, together with the other genetic causes of this disorder, such as ‘gain-of-function’ CDKN1C mutations12 and maternal 11p15 duplications.18 It is worth noting that the parent of origin-dependent dominant pedigrees are expected with CDKN1C mutations and 11p15 duplications but not with mutations of the non-imprinted HMGA2.
References
Eggermann, T., Gonzalez, D., Spengler, S., Arslan-Kirchner, M., Binder, G. & Schönherr, N. Broad clinical spectrum in Silver-Russell syndrome and consequences for genetic testing in growth retardation. Pediatrics 123, e929–e931 (2009).
Abu-Amero, S., Monk, D., Frost, J., Preece, M., Stanier, P. & Moore, G. E. The genetic aetiology of Silver-Russell syndrome. J. Med. Genet. 45, 193–199 (2008).
Menten, B., Buysse, K., Zahir, F., Hellemans, J., Hamilton, S. J., Costa, T. et al. Osteopoikilosis, short stature and mental retardation as key features of a new microdeletion syndrome on 12q14. J. Med. Genet. 44, 264–268 (2007).
Buysse, K., Reardon, W., Mehta, L., Costa, T., Fagerstrom, C., Kingsbury, D. J. et al. The 12q14 microdeletion syndrome: additional patients and further evidence that HMGA2 is an important genetic determinant for human height. Eur. J. Med. Genet. 52, 101–107 (2009).
Spengler, S., Schönherr, N., Binder, G., Wollmann, H. A., Fricke-Otto, S., Mühlenberg, R. et al. Submicroscopic chromosomal imbalances in idiopathic Silver-Russell syndrome (SRS): the SRS phenotype overlaps with the 12q14 microdeletion syndrome. J.Med.Genet. 47, 356–360 (2010).
Lynch, S. A., Foulds, N., Thuresson, A. C., Collins, A. L., Annerén, G., Hedberg, B. O. et al. The 12q14 microdeletion syndrome: six new cases confirming the role of HMGA2 in growth. Eur. J. Hum. Genet. 19, 534–539 (2011).
Takenouchi, T., Enomoto, K., Nishida, T., Torii, C., Okazaki, T., Takahashi, T. et al. 12q14 microdeletion syndrome and short stature with or without relative macrocephaly. Am. J. Med. Genet A 158A, 2542–2544 (2012).
Alyaqoub, F., Pyatt, R. E., Bailes, A., Brock, A., Deeg, C., McKinney, A. et al. 12q14 microdeletion associated with HMGA2 gene disruption and growth restriction. Am. J. Med. Genet A 158A, 2925–2930 (2012).
Netchine, I., Rossignol, S., Dufourg, M. N., Azzi, S., Rousseau, A., Perin, L. et al. 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell–Silver syndrome: Clinical scoring system and epigenetic–phenotypic correlations. J. Clin. Endocrinol. Metab. 92, 3148–3154 (2007).
Bartholdi, D., Krajewska-Walasek, M., Ounap, K., Gaspar, H., Chrzanowska, K. H., Ilyana, H. et al. Epigenetic mutations of the imprinted IGF2-H19 domain in Silver– Russell syndrome (SRS): Results from a large cohort of patients with SRS and SRSlikephenotypes. J. Med. Genet. 46, 192–197 (2009).
Singh, G. & Cooper, T. A. Minigene reporter for identification and analysis of cis elements and trans factors affecting pre-mRNA splicing. Biotechniques 41, 177–181 (2006).
Brioude, F., Oliver-Petit, I., Blaise, A., Praz, F., Rossignol, S., Le Jule, M. et al. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J. Med. Genet. 50, 823–830 (2013).
Zhou, X., Benson, K. F., Ashar, H. R. & Chada, K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 376, 771–774 (1995).
Ligon, A. H., Moore, S. D., Parisi, M. A., Mealiffe, M. E., Harris, D. J., Ferguson, H. L. et al. Constitutional rearrangement of the architectural factor HMGA2: a novel human phenotype including overgrowth and lipomas. Am. J. Hum. Genet. 76, 340–348 (2005).
Weedon, M. N., Lettre, G., Freathy, R. M., Lindgren, C. M., Voight, B. F., Perry, J. R. et al. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat. Genet. 39, 1245–1250 (2007).
Gorbenko del Blanco, D., de Graaff, L. C., Posthouwer, D., Visser, T. J. & Hokken-Koelega, A. C. Isolated GH deficiency: mutation screening and copy number analysis of HMGA2 and CDK6 genes. Eur. J. Endocrinol. 165, 537–544 (2011).
Mari, F., Hermanns, P., Giovannucci-Uzielli, M. L., Galluzzi, F., Scott, D., Lee, B. et al. Refinement of the 12q14 microdeletion syndrome: primordial dwarfism and developmental delay with or without osteopoikilosis. Eur. J. Hum. Genet l7, 1141e7 (2009).
Begemann, M., Spengler, S., Gogiel, M., Grasshoff, U., Bonin, M., Betz, R. C. et al. Clinical significance of copy number variations in the 11p15.5 imprinting control regions: new cases and review of the literature. J. Med. Genet. 49, 547–553 (2012).
Wakeling, E. L., Abu Amero, S., Alders, M., Bliek, J., Forsythe, E., Kumar, S. et al. Epigenotype-phenotype correlations in Silver-Russell syndrome. J. Med. Genet. 47, 760–768 (2010).
Acknowledgements
We thank the patients and families for their cooperation and Thomas A. Cooper for the gift of the RHCglo minigene vector. This research was supported by grants from Telethon-Italia grant no. GGP11122, Progetto Bandiera MIUR-CNR Epigenomica, Associazione Italiana Ricerca sul Cancro no. 12815 and FP7 MC-ITN INGENIUM project no. 290123 (AR) and P.O.R. Campania FSE 2007-2013, Progetto CREMe, CUP B25B09000050007 (MVC).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
De Crescenzo, A., Citro, V., Freschi, A. et al. A splicing mutation of the HMGA2 gene is associated with Silver–Russell syndrome phenotype. J Hum Genet 60, 287–293 (2015). https://doi.org/10.1038/jhg.2015.29
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.29
This article is cited by
-
Screening of patients born small for gestational age with the Silver-Russell syndrome phenotype for DLK1 variants
European Journal of Human Genetics (2021)
-
12q14.3 microdeletion involving HMGA2 gene cause a Silver-Russell syndrome-like phenotype: a case report and review of the literature
Italian Journal of Pediatrics (2020)
-
Contribution of gene mutations to Silver-Russell syndrome phenotype: multigene sequencing analysis in 92 etiology-unknown patients
Clinical Epigenetics (2020)
-
Cri-du-chat syndrome mimics Silver-Russell syndrome depending on the size of the deletion: a case report
BMC Medical Genomics (2018)
-
Periodic reanalysis of whole-genome sequencing data enhances the diagnostic advantage over standard clinical genetic testing
European Journal of Human Genetics (2018)
