
Abstract
WWOX has been recently implicated in autosomal recessive spinocerebellar ataxia type 12 (SCAR12) and severe early-onset epileptic encephalopathy (EOEE). By array comparative genomic hybridization, we identified a 0.6 Mb homozygous deletion in 16q23.1 in a fetus presenting with brain anomalies. His older sister who died at the age of 22 months from an EOEE was also homozygous for the copy number variations in 16q23.1. This deletion includes the first six exons of WWOX and results in a null genotype in homozygous patients. This family gives additional support for the implication of WWOX in severe EOEEs. We report for the first time prenatal ultrasound findings in a fetus with a WWOX-null genotype. Our study expands the range of brain abnormalities in WWOX-related EOEEs. This additional family confirms the genotype–phenotype correlation with WWOX-null alleles associated with the most severe form of WWOX-related epileptic encephalopathy with premature death.
Similar content being viewed by others
Main
Germline biallelic missense mutations in the WWOX gene have recently been reported to cause a new autosomal recessive syndrome associating cerebellar ataxia with epilepsy and intellectual disability in two consanguineous families.1 In two other families, homozygous early premature termination codons in WWOX resulted in growth retardation, microcephaly, epileptic seizures, ophthalmological abnormalities (retinopathy and optic atrophy) and premature death in the first years of life.2, 3 Very recently, Mignot et al.4 reported five additional families with a WOREE syndrome (WWOX-related epileptic encephalopathy), a clinical entity similar to that reported in patients with early premature termination codons.2, 3 It seems there is a genotype/phenotype correlation for WWOX-related encephalopathies: null genotypes are associated with severe early-onset epileptic encephalopathies whereas hypomorphic missense mutations result in SCAR12. We report an additional family with severe early-onset epileptic encephalopathy and biallelic alterations of WWOX resulting in a null genotype.
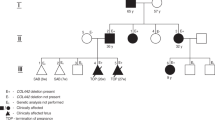
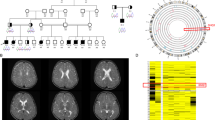
We describe a consanguineous family of Turkish origin (Figure 1a) with a WOREE syndrome. The molecular diagnosis of WWOX-related encephalopathy was made for the first time in the prenatal period after termination of pregnancy in a fetus with brain anomalies. The 34-year-old mother (I.2) was referred to our prenatal diagnosis department at the 35 weeks of gestation. It was the eighth pregnancy of this couple with three miscarriages, one boy with a right hand skeletal anomaly and hypospadias (II.1), and a girl who died prematurely at 22 months of obstructive cardiomyopathy with early-onset epileptic encephalopathy of unknown etiology (II.2). For the fetus II.5, the mother noted abnormal rhythmic kicks that did not appear to correspond to normal fetal movements during the third trimester of pregnancy and could suggest intrauterine seizures. Ultrasound scans and magnetic resonance imaging showed macrocephaly secondary to increased subarachnoid spaces with no ventriculomegaly, thin corpus callosum and bilateral frontal pseudocysts (Figure 2A). Nuchal lucency was normal on early ultrasound scans in II.5. The family chose not to continue the pregnancy. Post-mortem evaluation of the fetus at 39 weeks of pregnancy showed slight overgrowth, placental hypertrophy and organomegaly. Growth parameters were compatible with a 41 weeks gestation: weight was 3760 g (+2 s.d.), height 51.5 cm (+0.5 s.d.), head circumference 35.5 cm (+2.5 s.d.) and biparietal diameter 109 cm (+3 s.d.). Autopsy identified polymicrogyria that was only mentioned after fetal brain imaging, enlarged sylvian fissures and a thin corpus callosum (Figure 2C).

Family Pedigree and cytogenomic characterization of the 16q23.1 homozygous deletion. (a) Family pedigree and (b) genetic investigations: (a) aCGH analysis showing the deletion of the 5′ end of WWOX gene, (c) confirmation of the WWOX copy number variations by quantitative PCR on genomic DNAs from I.2 (green bars), I.1 (blue bars), II.2 and II.5, and a diploid control (red bars). Both parents (I.1 and I.2) are heterozygous whereas II.2 and II.5 are homozygous for the WWOXΔex 1–6 allele. A full color version of this figure is available at the Journal of Human Genetics journal online.

Brain magnetic resonance imaging (MRI) and neuropathological findings in affected subjects. (A) Brain MRI T2-weighted scan of II.5 at 38 weeks of gestation: a) midline sagittal slice showing a thin corpus callosum (black arrows), (b) coronal slice showing germinolysis paracysts (black arrows), large pericerebral fluid spaces (black arrow heads), opercular malformation with exposure of the insulae (white arrow heads) and (c) lateral sagittal slice showing opercular malformation with exposure of the insulae (white arrow head) and large pericerebral fluid spaces (black arrow). (B) Brain MRI of patient II.2 at 3 months. (a) T1-weighted sagittal slice showing a thin corpus callosum (white arrows), (b) T2-weighted axial slice showing cerebral white matter atrophy, opercular malformation with exposure of the insulae (white arrows head) and (c) T2-weighted axial slice showing large pericerebral fluid spaces (white arrow heads). (C) Midsagittal view of the brain of II.5 showing the polymicrogyria (black arrow heads) and the thin corpus callosum (black arrow). A full color version of this figure is available at the Journal of Human Genetics journal online.
Patient II.2 was born after a normal delivery at 37 weeks of gestation. Ultrasound examination had revealed increased nuchal translucency (>95th percentile) and a single umbilical artery in prenatal period. Fetal karyotype was normal (46,XX). Level III ultrasound scan was not performed in II.2. Birth growth parameters and extra-uterine adaptation were normal. She was transferred in a neonatal intensive care unit because of episodes of cyanosis in the first hours of life. Spastic hypertonia and convulsions started at the age of 3 months. Myoclonic movements and severe limb hypertonia were noticed. At this stage, electroencephalogram examinations were normal. Tonic spasms with axial hyperextension and elevation–flexion of upper limbs were noted at 2 months of age. Eye contact was absent. Electroencephalogram revealed generalized epileptic anomalies with hypsarrhythmia and slow spikes and spike-wave discharges. A West syndrome was diagnosed. Seizures partially responded to valproate, vigabatrine, high doses of hydrocortisone and clobazam. She did not acquire any developmental milestones. Later on, electroencephalogram readings were particularly disorganized with slow background activity. Brain magnetic resonance imaging showed brain atrophy, sylvian fissures enlargement and increased subarachnoid spaces mainly in frontal areas (Figure 2B). Magnetic resonance imaging found no evidence of polymicrogyria in II.2. At the age of 6 months, she presented with a cardiomyopathy and a scoliosis. At 19 months, clinical examination showed a worrying neurological development with regular seizures, severe hypertonia and absent eye contact. Weight was 7.180 kg (−3 s.d.), height 70.5 cm (−3 s.d.) and head circumference 43.5 cm (−2.5 s.d.) demonstrating acquired growth retardation that started at 9 months. II.2 died prematurely from obstructive cardiomyopathy. Investigations found no evidence of a storage disorder or a neurodegenerative metabolic disorder. Mitochondrial and lysosomal functions appeared to be normal.
Genetic investigations in II.2 by conventional karyotyping, aCGH with a first generation BAC/PAC array and molecular analyses of ARX/CDKL5 were normal. By using aCGH with a 180 K SurePrint G3 Human CGH Microarray Kit (Agilent, Santa Clara, CA, USA) in II.5 after termination of pregnancy, we identified a homozygous deletion in 16q23.1 with a minimal physical size of 594 kb (hg19; chr16:78 084 187–78 424 234) including the first six exons of WWOX (WWOXΔex 1–6; Figure 1b). A retrospective molecular diagnosis of WOREE syndrome was made for II.2. The elder sister was also homozygous for the WWOXΔex 1–6 allele. Quantitative PCR analysis demonstrated that both parents are heterozygous for the WWOXΔex 1–6 allele (Figure 1c).
We report the first case of severe WWOX-related phenotype diagnosed at a fetal stage after termination of pregnancy for brain anomalies at 33 weeks of gestation. II.5 and his affected sibling were shown to be homozygous for a deletion removing the 5′ end of WWOX. The WWOXΔex 1–6 allele is predicted to lead to a nonfunctional gene at the transcriptional level. In our family, both patients have a null genotype resulting in the complete absence of the WWOX protein. Patients with a null genotype2, 3, 4 are affected with a similar severe condition consisting of growth retardation, early-onset epileptic seizures, profound psychomotor delay, brain and ophthalmological anomalies and premature death (Table 1). Hypomorphic missense alleles are responsible for a less severe SCAR12 phenotype with increased life expectancy.1 Notably, parents have experienced three miscarriages that could also result from WWOX invalidation. Our data suggest an intrafamilial clinical heterogeneity for WWOX-null genotypes with life expectancies in affected individuals ranging from death at the early stages of prenatal development to premature death in the first years of life. Interestingly, the proband’s brother (II.1) presented with a right hand skeletal anomaly and hypospadias. Before this study, a WWOX deleterious allele (in-frame deletion of exons 6–8) was found in the heterozygous state in a male patient with gonadal dysgenesis.5 We could hypothesize that haploinsufficiency for WWOX is responsible for skeletal and genital abnormalities in II.1. WWOX genetic screen was not possible in II.1.
Altogether, 15 different germline mutations have been described in WWOX in 10 families with three missenses,1, 4 four premature termination codons2, 4 and eight copy number variations3, 4, 5 (and this study). Notably, large rearrangements account for more than 50% of all deleterious alleles. We wish to stress the importance of seeking a second deleterious allele in WWOX in all patients with a neurologic disorder who are heterozygous for an inherited WWOX-alteration identified either by aCGH or by whole exome sequencing.
In conclusion, our family gives additional support for the implication of WWOX in severe early-onset epileptic encephalopathies and possibly in disorder of sex development. It expands the phenotypic spectrum of WWOX-related phenotype with early fetal loss and brain anomalies detected in the prenatal period.
References
Mallaret, M., Synofzik, M., Lee, J., Sagum, C. A., Mahajnah, M. & Sharkia, R. et al. The tumour suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Brain 137, 411–419 (2014).
Abdel-Salam, G., Thoenes, M., Afifi, H. H., Korber, F., Swan, D. & Bolz, H. J. The supposed tumor suppressor gene WWOX is mutated in an early lethal microcephaly syndrome with epilepsy, growth retardation and retinal degeneration. Orphanet J. Rare Dis. 9, 12 (2014).
Ben-Salem, S., Al-Shamsi, A. M., John, A., Ali, B. R. & Al-Gazali, L. A novel whole exon deletion in WWOX gene causes early epilepsy, intellectual disability and optic atrophy. J. Mol. Neurosci. e-pub ahead of print 18 November 2014; doi:10.1007/s12031-014-0463-8.
Mignot, C., Lambert, L., Pasquier, L., Bienvenu, T., Delahaye-Duriez, A. & Keren, B. et al. WWOX-related encephalopathies: delineation of the phenotypical spectrum and emerging genotype-phenotype correlation. J. Med. Genet. 52, 61–70 (2015).
White, S., Hewitt, J., Turbitt, E., van der Zwan, Y., Hersmus, R. & Drop, S. et al. A multi-exon deletion within WWOX is associated with a 46,XY disorder of sex development. Eur. J. Hum. Genet. 20, 348–351 (2012).
Acknowledgements
We thank the family who participated in this study. We gratefully acknowledge Profs E Raffo and F Feillet for clinical investigations conducted in this family. We also thank A Saunier and Aïssa Mostaine for WWOX molecular screening.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Valduga, M., Philippe, C., Lambert, L. et al. WWOX and severe autosomal recessive epileptic encephalopathy: first case in the prenatal period. J Hum Genet 60, 267–271 (2015). https://doi.org/10.1038/jhg.2015.17
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.17
This article is cited by
-
WWOX-associated encephalopathies: identification of the phenotypic spectrum and the resulting genotype-phenotype correlation
Neurological Sciences (2018)
-
W44X mutation in the WWOX gene causes intractable seizures and developmental delay: a case report
BMC Medical Genetics (2016)
-
Novel mutations in WWOX, RARS2, and C10orf2 genes in consanguineous Arab families with intellectual disability
Metabolic Brain Disease (2016)
-
WWOX dysfunction induces sequential aggregation of TRAPPC6AΔ, TIAF1, tau and amyloid β, and causes apoptosis
Cell Death Discovery (2015)
