
Abstract
SOFT syndrome (MIM614813) is an extremely rare primordial dwarfism characterized by short stature, onychodysplasia, facial dysmorphism and hypotrichosis, which is caused by biallelic mutations in the POC1A gene. Only 19 patients with mutation-confirmed SOFT syndrome have been reported to date, all of whom carried homozygous variants that were strongly associated with consanguineous marriages. We report an 8.5-year-old boy with SOFT syndrome showing primordial dwarfism, no effect of growth-hormone therapy and skeletal dysplasia. This is the first report of compound heterozygous variants in POC1A, one previously reported and the other novel. A characteristic skeletal manifestation is reported.
Similar content being viewed by others


Introduction
SOFT (short stature, onychodysplasia, facial dysmorphism and hypotrichosis; MIM614813) syndrome is a rare genetic disorder that presents with primordial dwarfism, with a final height ranging from 112 to 127 cm (Z=−9.0 to −6.0).1 The facial dysmorphism includes a triangular face with a pointed chin, relative macrocephaly with frontal bossing, frontal balding and midface hypoplasia. In these patients, onychodysplasia presents as short rectangular fingers, particularly in distal phalanges and hypoplastic fingernails. The syndrome inherits as an autosomal recessive trait, and homozygous mutations in POC1A were identified as being causative.2, 3 To date, 19 patients with molecularly confirmed homozygous mutations inherited from nine consanguineous parents have been reported.2, 3, 4, 5, 6 Here, we report a Korean patient with SOFT syndrome harboring compound heterozygous POC1A mutations, which were detected by whole-exome sequencing, who presented with profound short stature without any effect of long-term growth-hormone therapy.
Case report
Written informed consent to participate in this study was obtained from the patient and his parents. Our Institutional Review Board approved the study.
The patient was born after an uncomplicated pregnancy as a first baby of healthy nonconsanguineous Korean parents. His father was 175 cm (Z=+0.3) and his mother was 173 cm (Z=+2.5) tall. He was born at 37+0 weeks of gestation with a birth weight of 1.66 kg (Z=−4.3) and a birth length of 39.0 cm (Z=−4.1). Prenatal ultrasonography performed around the 28th week of gestation suspected a short length for both upper and lower extremities, and he was cared for at the neonatal intensive care unit for 1 month. No congenital anomaly was detected in the heart, brain and intra-abdominal organs. A chromosome analysis revealed a normal male karyotype, 46, XY.
At the age of 22 months, his height was 61.9 cm (Z=−7.9) and his weight was 4.5 kg (Z=−8.9). His growth velocity was 7.6 cm per year (Z=−0.6) during the following year. Korean Infant and Child Development Test performed at the age of 24 months showed an appropriate social–personal, language and cognitive–adaptive development for his age, although gross and fine motor development was delayed for about 6 months compared to his peers. Growth-hormone stimulation tests showed normal hormonal responses. Under the clinical diagnosis of Silver–Russell syndrome (MIM180860), recombinant growth-hormone therapy was started at the age of 32 months, with a mean dosage of 0.3 mg kg−1 per week. However, his response to growth-hormone therapy was poor (4.6 cm per year, Z=−1.5); thus, its administration was discontinued after 5.3 years and the patient was referred to a genetics clinic (Figure 1).

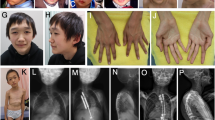
(a, b) The patient displays facial dysmorphism, including dolicocephaly, a triangular face with a pointed chin, sparse hair, brachydactyly and nail hypoplasia. (c) The patient’s growth chart shows the presence of severe short stature beyond the range of the genetic target height (TH). No effect of growth-hormone therapy lasting for 5.3 years was observed. A full color version of this figure is available at the Journal of Human Genetics journal online.
At the age of 8.5 years, his height was 96.5 cm (Z=−6.7), his arm span was 92 cm, weight was 12.6 kg (Z=−6.0) and head circumference was 48.3 cm (Z=−2.3). Generalized dark-pigmented skin, mild frontal bossing, a triangular face with a pointed chin, sparse hair, crowded teeth, short limbs and brachydactyly with short digital phalanges were observed. He wore spectacles because of hyperopia. A skeletal survey showed short long bones without modeling defects, hypoplastic iliac bones, short femoral necks with metaphyseal irregularity, mild flat epiphyses at the knees, short phalanges with distinct metaphyseal cupping and cone-shaped epiphyses, and tall vertebrae (Table 1 and Figure 2).

(a) At 2.7 years of age, before the onset of growth-hormone therapy, the left hand shows all short phalanges with minimal cupping at the phalangeal bases. The carpals are remarkably small and the estimated bone age is around 6 months. (b) At age 8.6 years, the left hand shows short middle and distal phalanges with apparent metaphyseal cupping and irregularity with cone-shaped epiphyses. The carpal bone age is around 7 years. (c) A radiograph of the pelvis shows the narrow and vertical appearance of the iliac bone, shallow acetabuli and short femoral necks with metaphyseal irregularity. (d) Teleradiograph of the lower extremity shows metaphyseal flaring at the knees with mild epiphyseal dysplasia and slender diaphyses of the long bones. Note the disproportionately short fibula. (e) A lateral view of the spine reveals distinct tall vertebrae.
Whole-exome sequencing was performed in the patient and his parents. The experimental processes were as described previously,7, 8 with minor modifications. Sequence variants were filtered based on the assumption of a recessive inheritance or a de novo dominant inheritance pattern. One novel c.239C>T, p.Ser80Phe sequence variant and one previously reported c.241C>T, p.Arg81* sequence variant in POC1A were identified in the proband (Figure 3a).3 The missense variant was expected to be damaging based on the functional prediction softwares (PhyloP: 5.433; Polyphen-2: 1.00; SIFT: 0.00). The variants were confirmed by Sanger sequencing, and the two variants found in the patient were independently inherited from his parents (Figure 3a). The variants were not listed in the 1000 Genomes, ExAC (http://exac.broadinstitute.org) or the National Heart, Lung and Blood Institute whole-exome database. Moreover, they were not discovered in 2040 chromosomes from the Korean population. The novel missense variant, p.Ser80Phe, is located in the WD40 domain of the protein and is a well-conserved amino-acid residue during evolution, which suggests functional implications for the change (Figure 3b).

(a) Pedigrees of the family carrying compound heterozygous POC1A mutations, as confirmed by Sanger sequencing. Affected and unaffected individuals are denoted by black and white symbols, respectively. POC1A alleles are represented by ‘WT’ (wild type), ‘c.239C>T’ (p.Ser80Phe, missense mutation) and ‘c.241C>T’ (p.Arg81*, nonsense mutation). (b) Secondary POC1A protein structure with compound heterozygous mutations and a known variant. Amino-acid conservation of the POC1A Ser80Phe residue in orthologs from different vertebrate species. B.t., Bos taurus; D.r., Danio rerio; G.g., Gallus gallus; H.s., Homo sapiens; M.m., Mus musculus; O.c., Oryctolagus cuniculus; X.l., Xenopus laevis.
Discussion
We identified biallelic mutations in the POC1A gene in a patient with a clinical phenotype of SOFT syndrome. POC1A encodes the POC1 centriolar protein A, which plays a role in centrosome-mediated cell mitosis control via mitotic spindle organization and cilia formation.9 Therefore, SOFT syndrome could be classified as a type of ciliopathy.10 The novel missense variant identified in this study is located in the WD40 domain of POC1A. The WD40 domain is necessary to localize the protein to centrioles, and defective WD40 domain will inhibit the proper protein localization.9 Therefore, we hypothesize that p.Ser80Phe variant may induce aberrant POC1A localization and interrupt with the normal function of the protein.
The primordial dwarfism and characteristic facial features observed in patients with SOFT syndrome might overlap with those of Silver–Russell syndrome. However, children with Silver–Russell syndrome benefit from growth-hormone supplementation, even in the absence of growth-hormone deficiency, regarding both significant growth acceleration and improved final height,11 whereas patients with SOFT syndrome do not respond to growth-hormone supplementation at all,6 which was also observed in our patient. SOFT syndrome has unique skeletal findings. Rhizomelic short upper and lower limbs and delayed ossification of the proximal humeral and femoral epiphyses are noticeable during infancy and childhood, but become less evident with age. Other findings include a dysplasic acetabulum with short and thick femoral necks, tall vertebrae and short metacarpal and phalangeal bones with cone-shaped epiphyses.1, 4, 5, 12 As differential diagnoses, other skeletal dysplasias showing short limbs and sparse hair such as cranioectodermal dysplasia (MIM218330) and cartilage-hair hypoplasia (MIM250250)-anauxetic dysplasia (MIM607095) spectrum should be considered.13, 14 Unlike SOFT syndrome, cranioectodermal dysplasia frequently accompanies nephronophthisis, craniosynostosis and dental anomalies,14 and cartilage-hair hypoplasia-anauxetic dysplasia spectrum is usually associated with immunodeficiency, Hirschsprung disease, malignancies or autoimmune diseases.13 In SOFT syndrome, no known intrinsic abnormality has been reported.1, 2, 3
In summary, we report the first patient with SOFT syndrome harboring compound heterozygous variants of POC1A. In patients presenting with primordial short stature, a meticulous radiological analysis especially focused on the hands, hips and spine may give alertness to the diagnosis of SOFT syndrome. Moreover, understanding POC1A mutations may provide appropriate management and genetic counseling to these patients and their families.
References
Shalev, S. A., Spiegel, R. & Borochowitz, Z. U. A distinctive autosomal recessive syndrome of severe disproportionate short stature with short long bones, brachydactyly, and hypotrichosis in two consanguineous Arab families. Eur. J. Med. Genet. 55, 256–264 (2012).
Sarig, O., Nahum, S., Rapaport, D., Ishida-Yamamoto, A., Fuchs-Telem, D., Qiaoli, L. et al. Short stature, onychodysplasia, facial dysmorphism, and hypotrichosis syndrome is caused by a POC1A mutation. Am. J. Hum. Genet. 91, 337–342 (2012).
Shaheen, R., Faqeih, E., Shamseldin, H. E., Noche, R. R., Sunker, A., Alshammari, M. J. et al. POC1A truncation mutation causes a ciliopathy in humans characterized by primordial dwarfism. Am. J. Hum. Genet. 91, 330–336 (2012).
Chen, J. H., Segni, M., Payne, F., Huang-Doran, I., Sleigh, A., Adams, C. et al. Truncation of POC1A associated with short stature and extreme insulin resistance. J. Mol. Endocrinol. 55, 147–158 (2015).
Koparir, A., Karatas, O. F., Yuceturk, B., Yuksel, B., Bayrak, A. O., Gerdan, O. F. et al. Novel POC1A mutation in primordial dwarfism reveals new insights for centriole biogenesis. Hum. Mol. Genet. 24, 5378–5387 (2015).
Barraza-Garcia, J., Ivan Rivera-Pedroza, C., Salamanca, L., Belinchon, A., Lopez-Gonzalez, V., Sentchordi-Montane, L. et al. Two novel POC1A mutations in the Primordial dwarfism, SOFT syndrome: Clinical homogeneity but also unreported malformations. Am. J. Med. Genet. A 170, 210–216 (2016).
Choi, M., Scholl, U. I., Ji, W. Z., Liu, T. W., Tikhonova, I. R., Zumbo, P. et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl Acad. Sci. USA 106, 19096–19101 (2009).
Goh, G. & Choi, M. Application of whole exome sequencing to identify disease-causing variants in inherited human diseases. Genomics Inform. 10, 214–219 (2012).
Pearson, C. G., Osborn, D. P., Giddings, T. H. Jr, Beales, P. L. & Winey, M. Basal body stability and ciliogenesis requires the conserved component Poc1. J. Cell Biol. 187, 905–920 (2009).
Tobin, J. L. & Beales, P. L. The nonmotile ciliopathies. Genet. Med. 11, 386–402 (2009).
Azcona, C., Albanese, A., Bareille, P. & Stanhope, R. Growth hormone treatment in growth hormone-sufficient and -insufficient children with intrauterine growth retardation/Russell–Silver syndrome. Horm. Res. 50, 22–27 (1998).
Turnpenny, P. D. & Thwaites, R. J. Dwarfism, rhizomelic limb shortness, and abnormal face: new short stature syndrome sharing some manifestations with Robinow syndrome. Am. J. Med. Genet. 42, 724–727 (1992).
Rider, N. L., Morton, D. H., Puffenberger, E., Hendrickson, C. L., Robinson, D. L. & Strauss, K. A. Immunologic and clinical features of 25 Amish patients with RMRP 70A—>G cartilage hair hypoplasia. Clin Immunol. 131, 119–128 (2009).
Amar, M. J., Sutphen, R. & Kousseff, B. G. Expanded phenotype of cranioectodermal dysplasia (Sensenbrenner syndrome). Am. J. Med. Genet. 70, 349–352 (1997).
Acknowledgements
This study was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI12C0014).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Min Ko, J., Jung, S., Seo, J. et al. SOFT syndrome caused by compound heterozygous mutations of POC1A and its skeletal manifestation. J Hum Genet 61, 561–564 (2016). https://doi.org/10.1038/jhg.2015.174
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.174
This article is cited by
-
Identification of SOFT syndrome caused by a pathogenic homozygous splicing variant of POC1A: a case report
BMC Medical Genomics (2021)
-
Microcephalic Osteodysplastic Primordial Dwarfism, Type II: a Clinical Review
Current Osteoporosis Reports (2017)
