
Abstract
Brachydactyly refers to shortening of digits due to hypoplasia or aplasia of bones forming the hands and/or feet. Isolated brachydactyly type E (BDE), which is characterized by shortened metacarpals and/or metatarsals, results in a small proportion of patients from HOXD13 or PTHLH mutations, although in the majority of cases molecular lesion remains unknown. BDE, like other brachydactylies, shows clinical heterogeneity with highly variable intrafamilial and interindividual expressivity. In this study, we investigated two Polish cases (one familial and one sporadic) presenting with BDE and additional symptoms due to novel PTHLH mutations. Apart from BDE, the affected family showed short stature, mild craniofacial dysmorphism and delayed bone age. Sanger sequencing of PTHLH revealed a novel heterozygous frameshift mutation c.258delC(p.N87Tfs*18) in two affected individuals and one relative manifesting mild brachydactyly. The sporadic patient, in addition to BDE, presented with craniofacial dysmorphism, normal stature and bone age, and was demonstrated to carry a de novo heterozygous c.166C>T(p.R56*) mutation. Our paper reports on the two novel truncating PTHLH variants, resulting in variable combination of BDE and other symptoms. Data shown here expand the knowledge on the phenotypic presentation of PTHLH mutations, highlighting significant clinical variability and incomplete penetrance of the PTHLH-related symptoms.
Similar content being viewed by others

Introduction
Brachydactyly type E (BDE; MIM#113300, #613382) is characterized by variable shortening of the metacarpals and/or metatarsals. The majority of BDE cases represent a syndromic manifestation of the defect, such as Turner syndrome, 2q37.3 deletion syndrome and Albright hereditary osteodystrophy.1, 2 Hertzog classified BDE into three distinct entities, that is, type E1—with shortening of IVth metacarpals (sometimes with short metatarsals), type E2—with shortening of metacarpals IV and V, and/or metatarsals, including shortening of the distal phalanx of the thumb and type E3—with variable combinations of short metacarpals, without phalangeal involvement.3 BDE, like other brachydactylies, shows high intrafamilial and interindividual clinical heterogeneity, with variable expressivity of the phenotypic features. Isolated BDE or BDE with short stature is inherited as an autosomal dominant trait and in a small subset of patients is caused by heterozygous HOXD13 (MIM 142989) or PTHLH (MIM 168470) mutations, although in the majority of cases the underlying genetic defect remains unknown.4, 5, 6, 7, 8, 9
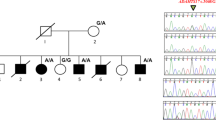
In this study, we investigated a two-generation family (Figure 1a) and an unrelated sporadic patient (Figure 1d) of Polish origin, both presenting with BDE and other symptoms caused by PTHLH mutations.

(a) The pedigree of the presented family. Individuals available for clinical and genetic testing are indicated with plus symbol. (b) Lateral view of the index (patient III-3) showing prominent forehead/frontal bossing, flat facial profile, small nose with depressed nasal bridge and anteverted nares. (c) Lateral view of the patient III-1 manifesting flat facial profile and upturned nose with slightly depressed nasal bridge. (d) The pedigree of the sporadic case. (e) Lateral view of the sporadic patient (II-1′) showing upturned nose and slightly depressed nasal bridge. (f) Schematic localization of all known intragenic mutations in the PTHLH gene. Mutations previously described in the literature are shown in black,6, 16 whereas two variants reported here are depicted in red. Green boxes represent 5′- and 3′-UTRs, whereas blue boxes depict protein coding sequence. Introns are shown in gray. A full color version of this figure is available at the Journal of Human Genetics journal online.
Results
Familial case
Patient III-3
The index (patient III-3) was a female individual born to a non-consanguineous, healthy couple. Her body measurements at the age of 4.5 years revealed short stature with height 100 cm (<3rd percentile), body weight 16.5 kg (10th–25th percentile) and head circumference (HC) 53 cm (90th–97th percentile). Dysmorphological assessment showed facial dysmorphism, (Figure 1b) and generalized brachydactyly of the hands and feet (Figures 2a and b). Radiological evaluation demonstrated BDE (Figure 2c) and delayed bone age (1.5 years delay at the age of 5). Furthermore, the index had thoracic scoliosis and lumbar hyperlordosis (Figures 2d and e). Karyotype, endocrine studies and laboratory tests of the bone metabolism were unremarkable. Psychomotor development was normal. The patient was reassessed at the age of 6 years. Interestingly, she was no longer short statured with height 109.5 cm (3rd–10th percentile); however, relatively large cranium (Figure 2f) with HC of 54 cm (>97th percentile) was observed.

Clinical and radiological features observed in the index patient (III-3) at the age of 5 years (Figures a–f) as well as in the sporadic index case (II-1') at the age of 13 years (Figures g–i). (a) Brachydactyly of hands most pronounced in fingers 2 and 5, with clinically observed shortening of the distal phalanges. (b) Brachydactyly of feet characterized by broad, stocky feet, short toes 1–4 due to apparent shortening of the distal phalanges; wide and valgus halluces. (c) Hand radiograph showing short and stubby metacarpals most pronounced in bones I and V, and short middle phalanx of the index finger (indicated with white arrows). In addition, short terminal phalanges and abnormal cone-shaped epiphyses of the metacarpals, as well as delayed bone age were noted. (d, e) X-ray of the vertebral column revealing thoracic scoliosis and lumbar hyperlordosis. (f) X-ray of the head showing large cranium in relation to the face. (g) Brachydactyly of hands most pronounced in fingers 2, 3 and 5. (h) Radiograph of left hand presenting short metacarpals IV and V, as well as shortened middle and terminal phalanges most evident in fingers 2, 3 and 5. (i) Brachydactyly of feet characterized by broad and stocky feet, short toes 3–5 due to shortening of the corresponding metatarsals; wide and valgus halluces. A full color version of this figure is available at the Journal of Human Genetics journal online.
Patient III-1
The patient (III-1) presented with facial dysmorphism (Figure 1c) and growth retardation since 6 years of age. Her body measurements at the age of 9 years were following: height 124 cm (3rd percentile), body weight 26.8 kg (25th–50th percentile) and HC 53 cm (50th–75th percentile). There was recognizable BDE in both hands (Figures 3a and b) and feet (Figures 3c and d), and bone age was delayed by 3 years. The patient manifested hypothyroidism with elevated anti-TPO antibodies. Her intellectual development was normal. Karyotype was normal.

Clinically and radiologically variable presentation of BDE in the proband’s III-3 sister, that is, patient III-1 at the age of 9 years (a–d) and the father (patient II-1) aged 47 years (e–h). (a) Hands presenting with length reduction of 5th fingers and thumbs in both hands. (b) X-ray of the hands showing shortened and stubby metacarpals I, III–V and short middle phalanges of fingers 2 and 5 (designated by white triangles), as well as abnormal cone-shaped epiphyses of metacarpals prematurely fused to their metaphyses. (c) Feet presenting with apparently short toes 3–5 and prominent sandal gaps. (d) X-ray of the feet showing typical BDE symptoms, that is, short metatarsals III–V. (e) Discrete brachydactyly comprising short and stubby thumbs in otherwise clinically normal hands. (f) Hand X-ray disclosing short first metacarpals and mild shortening of the middle phalanges of fingers 2 and 5 (marked with white triangles). (g) Mild shortening of the post-axial rays in both feet (digits 3–5). (h) Feet X-ray showing hypoplastic middle and distal phalanges of toes 3–5 (indicated by white arrows) and less prominent shortening of metatarsals 4–5. A full color version of this figure is available at the Journal of Human Genetics journal online.
Patient II-1
Patient II-1 was the 47-year old father of both sisters. The patient had mild presentation of brachydactyly, without short stature and only discrete limb anomalies (Figures 3e–h). His height was 184 cm (75th–90th percentile) and HC 58.5 cm (75th–90th percentile).
Sporadic patient
Patient II-1′ was a female individual born to an unrelated, healthy couple. At the age of 13 years, she manifested facial dysmorphism (Figure 1e) and BDE of the hands and feet (Figures 2g, h and i). Her body measurements were following: height 158 cm (25th–50th percentile), body weight 40.5 kg (10th–25th percentile) and HC 55 cm (50th–75th percentile). Bone age was relevant to metrical age. Throughout the childhood, the patient was never short statured and was intellectually normal. Karyotype was normal.
Sequencing results and expression studies
Genomic DNA and total RNA were isolated from peripheral blood leukocytes. Sanger sequencing of the entire coding portion of PTHLH (NM_198965.1) detected a heterozygous frameshift mutation c.258delC(p.N87Tfs*18) in exon 4 of PTHLH in proband III-3. Familial studies revealed that the mutation was identified only in the brachydactyly affected family members. Sporadic patient (II-1′) was demonstrated to carry a de novo heterozygous nonsense c.166C>T(p.R56*) mutation. Both PTHLH variants were not found in 200 ethnically matched controls and were not annotated in the 1000 Genomes Project, dbSNP144, ExAC, HGMD, and NHLBI EVS databases.10, 11, 12, 13, 14 Finally, using real-time PCR with SYBR Green and human GAPDH as a standard, we measured the PTHLH mRNA level in all affected individuals in reference to eight healthy controls. However, owing to low PTHLH expression in blood lymphocytes, the results were uninformative.
Discussion
Mutations in the PTHLH gene give rise to a specific pattern of skeletal anomalies referred to as ‘BDE with short stature, PTHLH type’ characterized by additional clinical features, such as oligodontia, delayed tooth eruption, premature closure of growth plates, and variable middle and distal phalangeal involvement.1, 6, 15 In 2010, Klopocki et al.6 described several pathogenic mutations of the PTHLH, including one microdeletion encompassing the gene and four intragenic point mutations (p.L44P, p.L60P, p.K120*, p.*178W) in five unrelated families affected by BDE and short stature. Recently, another family with BDE and short stature carrying p.L15R PTHLH variant has been reported in the literature.16 For schematic representation of all point mutations described in PTHLH, see Figure 1f. The family reported by us harbored a novel c.258delC(p.N87Tfs*18) heterozygous PTHLH frameshift mutation, resulting in BDE with short stature/growth deceleration and delayed bone age. Interestingly, all symptoms, although variable in expressivity, were noted in the two female siblings (aged 6 and 9 years), whereas their 47-year-old father showed only mild BDE phenotype and normal stature. To our knowledge, delayed bone age has not been described in the PTHLH-related BDE patients, therefore we suggest that this may be an additional clinical feature attributed to the PTHLH loss of function mutations. The sporadic patient who carried a novel heterozygous c.166C>T(p.R56*) PTHLH mutation, manifested however BDE in association with normal stature and bone age. Both mutations introduce premature stop codons in the middle of PTHLH, thereby most probably giving rise to the transcript degradation via nonsense-mediated decay. Thus, haploinsufficiency is expected to occur in our families, consistently with the mutational pathomechanism proposed by Klopocki et al.6
In conclusion, delayed bone age has not been thus far reported as a clinical feature associated with PTHLH mutations. We therefore contribute here to the knowledge on the ‘BDE with short stature, PTHLH type’ related clinical symptoms, point to a significant phenotypic variability of the disease and expand the spectrum of pathogenic mutations of the PTHLH gene.
References
Mundlos, S. The brachydactylies: a molecular disease family. Clin. Genet. 76, 123–136 (2009).
Schwabe, G. C . & Mundlos, S. Genetics of Congenital Hand Anomalies. Handchir. Mikrochir. Plast. Chir. 36, 85–97 (2004).
Hertzog, K. P. Brachydactyly and pseudo-pseudohypoparathyroidism. Acta Genet. Med. Gemellol. 17, 428–438 (1968).
Caronia, G., Goodman, F. R., McKeown, C. M., Scambler, P. J. & Zappavigna, V. An I47L substitution in the HOXD13 homeodomain causes a novel human limb malformation by producing a selective loss of function. Development 130, 1701–1712 (2003).
Johnson, D., Kan, S. H., Oldridge, M., Trembath, R. C., Roche, P., Esnouf, R. M. et al. Missense mutations in the homeodomain of HOXD13 are associated with brachydactyly types D and E. Am. J. Hum. Genet. 72, 984–997 (2003).
Klopocki, E., Hennig, B. P., Dathe, K., Koll, R ., de Ravel, T., Baten, E. et al. Deletion and point mutations of PTHLH cause brachydactyly type E. Am. J. Hum. Genet. 86, 434–439 (2010).
Maass, P. G., Wirth, J ., Aydin, A., Rump, A., Stricker, S., Tinschert, S. et al. A cis-regulatory site downregulates PTHLH in translocation t(8;12)(q13;p11.2) and leads to Brachydactyly type E. Hum. Molec. Genet. 19, 848–860 (2010).
Brison, N ., Debeer, P., Fantini, S., Oley, C., Zappavigna, V., Luyten, F. P. et al. An N-terminal G11A mutation in HOXD13 causes synpolydactyly and interferes with Gli3R function during limb pre-patterning. Hum. Molec. Genet. 21, 2464–2475 (2012).
Jamsheer, A., Sowińska, A., Kaczmarek, L. & Latos-Bieleńska, A. Isolated brachydactyly type E caused by a HOXD13 nonsense mutation: a case report. BMC Med. Genet. 13, 4 (2012).
The 1000 Genomes Project. Available at http://www.1000genomes.org/. Accessed 9 December 2015.
The Database of Short Genetic Variation (dbSNP) National Center for Biotechnology Information. Available at http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi Accessed 9 December 2015.
The Exome Aggregation Consortium (ExAC). Available at http://exac.broadinstitute.org/. Accessed 9 December 2015.
The Human Gene Mutation Database. Available at http://www.hgmd.cf.ac.uk/ac/index.php. Accessed 9 December 2015.
NHLBI Exome Sequencing Project (ESP) Exome Varian Server (EVS). Available at http://evs.gs.washington.edu/EVS/. Accessed 9 December 2015.
Pereda, A., Garin, I ., Garcia-Barcina, M ., Gener, B., Beristain, E., Ibañez, A. M. et al. Brachydactyly E: isolated or as a feature of a syndrome. Orphanet J. Rare Dis. 8, 141 (2013).
Wang, J., Wang, Z., An, Y., Wu, C ., Xu, Y., Fu, Q. et al. Exome sequencing reveals a novel PTHLH mutation in a Chinese pedigree with brachydactyly type E and short stature. Clin. Chim. Acta. 446, 9–14 (2015).
Acknowledgements
We are grateful to the patients for participating in this study. This research was supported by the National Centre for Research and Development grant LIDER/008/431/L-4/12/NCBR/2013 to AJ and by the Polish National Science Centre grants UMO-2011-03-B-NZ5-00510 and UMO-2011-03-D-NZ2-06136 to AJ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Jamsheer, A., Sowińska-Seidler, A., Olech, E. et al. Variable expressivity of the phenotype in two families with brachydactyly type E, craniofacial dysmorphism, short stature and delayed bone age caused by novel heterozygous mutations in the PTHLH gene. J Hum Genet 61, 457–461 (2016). https://doi.org/10.1038/jhg.2015.172
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.172
This article is cited by
-
What to consider when pseudohypoparathyroidism is ruled out: iPPSD and differential diagnosis
BMC Medical Genetics (2018)
-
Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement
Nature Reviews Endocrinology (2018)
