
Abstract
A genome-wide association study (GWAS) was conducted to examine genetic associations of common autosomal nucleotide variants with sex in a Korean population with 4183 males and 4659 females. Nine genetic association signals were identified in four intragenic and five intergenic regions (P<5 × 10−8). Further analysis with an independent data set confirmed two intragenic association signals in the genes encoding protein phosphatase 1, regulatory subunit 12B (PPP1R12B, intron 12, rs1819043) and dynein, axonemal, heavy chain 11 (DNAH11, intron 61, rs10255013), which are directly involved in the reproductive system. This study revealed autosomal genetic variants associated with sex ratio by GWAS for the first time. This implies that genetic variants in proximity to the association signals may influence sex-specific selection and contribute to sex ratio variation. Further studies are required to reveal the mechanisms underlying sex-specific selection.
Similar content being viewed by others

Introduction
The total number of offspring born as female and as male individuals of a diploid population should theoretically be the same. In reality, however, the sex ratio of humans at birth has differed from one. For example, sex ratios of European populations have been fairly consistent, with the male-to-female ratio ranging from 1.02 to 1.08.1, 2, 3 Sex ratio can be affected by differential survival time of X-bearing and Y-bearing sperms in a variety of conditions including gonadotropins/testosterone concentration at conception, ovulation induction, paternal age, parity, birth order, coital rates, infertility, parental illness, maternal malnutrition/smoking/exposure to toxic agents and stress.2, 4, 5, 6, 7 Many genetic factors are suspected from the endogenous conditions. In fact, the sex ratio has been identified as paternally inherited in previous studies, in which men with more brothers had more sons and men with more sisters had more daughters.8, 9, 10 This may not be attributed to any Y chromosomal genes because the selection bias towards producing sons may be accelerated by constraining daughter-producing alleles from being inherited. Autosomal genes may be involved in sex ratio as shown in a simulation study where sex ratio was in equilibrium by frequency-dependent selection acting on the inheritance of polymorphic autosomal genes expressed in the male reproductive system.10
A meta-analysis across 51 genome-wide association studies (GWASs) with Caucasian populations was conducted to identify common autosomal variants for influencing sex ratio.11 The study, however, did not reveal any nucleotide variants significantly associated with sex (P>5 × 10−8). This might have resulted from the dilution of genetic effects by utilizing populations under different environmental conditions that interacted with genetic effects. We conducted a GWAS to examine genetic associations of common autosomal variants with sex in a large cohort of Koreans.
Materials and methods
Subjects and genotypes
This study used data collected from community-based cohorts by the Korean Association REsource (KARE) project. They included a total of 10 038 Korean individuals born from 1931 to 1963. Ethical approval was obtained from the Institutional Review Board of the Korea National Institute of Health, and all participants provided written informed consent after the aims and nature of the study were disclosed. All subjects were genotyped using the Affymetrix Genome-Wide Human SNP Array 5.0.12 Bayesian Robust Linear Modeling with the Mahalanobis Distance genotyping algorithm was used to obtain data.13 This study included 8842 subjects (4183 males and 4659 females) after screening for a genotype call rate smaller than 95%, sample contamination, gender inconsistency, cryptic relatedness and serious concomitant illness. A series of quality control procedures was also conducted to remove nucleotide variants with a subject call rate smaller than 95% or with minor allele frequency less than 0.05. Variants in Hardy–Weinberg disequilibrium (P<0.001) in both females and males were also excluded. This study used a total of 1 400 289 nucleotide variants, which included 1 118 622 variants imputed based on filtered haplotypes of 170 Japanese and Chinese HapMap founders (HapMap 3 phase 2; http://hapmap.ncbi.nlm.nih.gov/) as a reference panel.
Association analysis
Genetic associations were analyzed using a mixed model, which employed a genomic relationship matrix for random polygenic effects to avoid population stratification14 and incorporated the leaving-one-chromosome-out approach to avoid underestimation of genetic association15 as follows:

where Y is the vector of dichotomous phenotypes (sex); μ is the overall mean; 1 is the vector of 1's; β is the fixed effect for the minor allele of the candidate nucleotide variant; and x is the vector with elements of 0, 1 and 2 for the homozygote of the minor allele, heterozygote and homozygote of the major allele, respectively. g− is the vector of random polygenic effects  using the genome without the chromosome housing the candidate variant, where G− is the n × n genomic relationship matrix without one chromosome, and
using the genome without the chromosome housing the candidate variant, where G− is the n × n genomic relationship matrix without one chromosome, and  is the polygenic variance component that should be re-estimated whenever a specific chromosome is excluded from the calculation of the genomic relationship matrix. The genomic relationship matrix consists of pairwise genetic relationship coefficients, which were estimated using genotypes of variants in linkage equilibrium (r2 > 0.8) as follows:
is the polygenic variance component that should be re-estimated whenever a specific chromosome is excluded from the calculation of the genomic relationship matrix. The genomic relationship matrix consists of pairwise genetic relationship coefficients, which were estimated using genotypes of variants in linkage equilibrium (r2 > 0.8) as follows:

where Nc is the number of autosomes (=22), Nm is the number of variants in the ith autosome, Pij is the frequency of the minor allele at the jth variant in the ith autosome, and nijk (nijl) is the number (0, 1 or 2) of the minor allele at the jth variant in the ith autosome for the kth (lth) individuals. ɛ is the vector of random residuals  , where
, where  is the residual variance component, and I is the n × n identity matrix. The polygenic and residual variance components were estimated using restricted maximum likelihood. Polygenic and residual variance components were first estimated by expectation maximization-restricted maximum likelihood, and then the estimates were used as initial values to obtain their average information-restricted maximum likelihood estimates. The fixed effect was then solved with the estimated variance components under the mixed model equations. The statistical analysis was conducted using the Genome-wide Complex Trait Analysis (GCTA) freeware.16
is the residual variance component, and I is the n × n identity matrix. The polygenic and residual variance components were estimated using restricted maximum likelihood. Polygenic and residual variance components were first estimated by expectation maximization-restricted maximum likelihood, and then the estimates were used as initial values to obtain their average information-restricted maximum likelihood estimates. The fixed effect was then solved with the estimated variance components under the mixed model equations. The statistical analysis was conducted using the Genome-wide Complex Trait Analysis (GCTA) freeware.16
Linkage disequilibrium blocks were constructed to determine if some variants associated with sex were considered to be one association signal. The linkage disequilibrium blocks were estimated at association signals using Haploview.17
Replication analysis
Genetic associations with sex ratio were further examined using an independent Korean population to validate genetic associations revealed in the current GWAS. This included 935 subjects recruited from routine health checkups at Hallym University Hospital.18, 19 Genomic DNA was extracted from their peripheral blood cells using QIAamp DNA blood mini kit (Qiagen, Hilden, Germany). Nucleotide variants were genotyped using the TaqMan polymerase chain reaction assay (Applied Biosystems, Foster City, CA, USA). All reactions were carried out following the manufacturer’s protocol, and the products resulted from the reactions were analyzed using ABI PRISM 7900HT (Applied Biosystems). Genetic associations were determined by the Fisher’s exact test under the assumption of an additive contribution of minor allele. Multiple testing by Bonferroni correction was also conducted for the association analysis.
Results
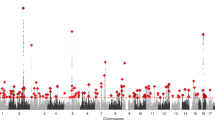
The genome-wide association analysis revealed 14 single-nucleotide polymorphisms associated with sex (P<5 × 10−8; Figure 1; Table 1). After excluding single-nucleotide polymorphisms in strong linkage disequilibrium (r2>0.8; Figure 2), nine association signals were identified for sex ratio distortion (Table 1). Five of the signals were observed with lower minor allele frequency in men than in women. They were all deviated from Hardy–Weinberg equilibrium (HWE) in men (P<10−3), but not in women (P>10−3) except for one signal (rs3013386), which showed a deviation in women (P<10−3), but not in men (P>10−3). The other four association signals were observed with lower minor allele frequency in women than in men. They were all deviated from HWE in women (P<10−3), but not in men (P>10−3).

Manhattan plot for genome-wide association with sex by autosomal position. Red horizontal line indicates genome-wide significance threshold value (P=5 × 10−8). A full color version of this figure is available at the Journal of Human Genetics journal online.

Linkage disequilibrium (LD) blocks with nucleotide variants in proximity of association signals for sex ratio. Pair-wise linkage disequilibrium estimate (r2) is presented in each cell. Red box indicates nucleotide variant with the most significant association within each LD block. (a) chromosome 1, (b) chromosome 10 and (c) chromosome 15. A full color version of this figure is available at the Journal of Human Genetics journal online.
Of the nine association signals, three were located in genes encoding protein phosphatase 1, regulatory subunit 12B (PPP1R12B, intron 12, rs1819043); dynein, axonemal, heavy chain 11 (DNAH11, intron 61, rs10255013); and ornithine aminotransferase (OAT, intron 6, rs11244715). The rs9788182 on chromosome 12 was an intronic variant within non-coding genes (LOC105369897, LOC105369897). The others were all intergenic variants.
Further genetic association analysis using an independent Korean population confirmed the associations of sex ratio with the intronic variants of PPP1R12B and DNAH11 genes (P<5.56 × 10−3; Table 2). The PPP1R12B variant was deviated from HWE in men (P<0.05), but not in women (P>0.05). The PPP1R12B variant was deviated from HWE in women (P<0.05), but not in men (P>0.05). These sex-specific Hardy–Weinberg disequilibrium were corresponding to the results from the genome-wide association analysis.
Discussion
The current GWAS identified nine common autosomal variants associated with sex ratio in a large cohort of Koreans. This implies sex-specific selections against particular common autosomal genetic variants. The mixed model analysis of the current study showed that autosomal genetic variants might explain 7.5% of phenotypic variability as polygenic effects. This implies that sex-specific selections against particular autosomal genetic variants may not be negligible. Furthermore, the associations of sex ratio with the intronic variants of DNAH11 and PPP1R12B genes were confirmed using another independent data set. The genes with variants that were discovered to be associated with sex ratio are potential candidates for sex ratio distortion. In particular, the genes encoding DNAH11 and PPP1R12B are involved in the reproductive system. The DNAH11 is used for the genetic diagnosis of patients with primary ciliary dyskinesia without ciliary ultrastructural defects,20 which show defects in the action of the flagella of sperm cells.21 The PPP1R12B engages in protein phosphorylation which might be influential in reproductive systems. Percoll fractionation analysis of human sperms showed that their morphology, motility, and hyperactivation were associated with protein tyrosine phosphorylation (P<0.05).22, 23 A gene enrichment analysis revealed protein phosphorylation to be associated with father-specific transmission distortion of a Yoruba population in Nigeria.24 Mouse sperm turned out to be functionally regulated by a gene (protein phosphatase 1 gamma 2, PP1γ2) with a role in protein phosphorylation.25 Furthermore, a considerably large amount of transcription of PPP1R12B was observed in the reproductive system.26
A caution should be addressed with regard to the association signals resulting from this study. We could not exclude the possibility that the associations may be involved in differential survival by sex after birth. This is because the current study dealt with the male-to-female sex ratio from ages in the 40s to 60s, but not at birth. Therefore, the results may reflect differential survival after birth to the age range examined, although we presumed that such a difference may not be considerable. Further investigations with sex ratio at birth are needed to distinguish differential survival before and after birth.
No genetic associations (P>5 × 10−8) were discovered with regard to sex ratio in a previous GWAS where a large-scale meta-analysis was conducted with 51 Caucasian cohorts.11 False-negative associations might be produced by the conservative multiple testing with the Bonferroni correction. Furthermore, confounding might be introduced when using multiple studies with different populations that had different genetic backgrounds under different environmental conditions. Also, the smaller sex ratio of Caucasians than that of Asians1 might make the study with Caucasians difficult to identify genetic associations.
The current association signals were identified from Korean populations. Nevertheless, this did not imply that the results are limited to only Koreans. Association studies with populations similar to Koreans (e.g., Japanese, Chinese and other Asians) would help enhance applicability of the signals.
Heterogeneous genetic variance of a particular trait by sex might be attributed partially to sex-specific selection against common genetic variants that are also associated with the trait. A larger minor allele frequency in one sex may produce larger genetic variance than in the other sex. Such heterogeneity would be reduced by excluding variants without HWE. For example, while genetic variances explained by nucleotide variants showed heterogeneity (P<0.05) by sex for body mass index, waist-to-hip ratio, pulse pressure, high-density lipoprotein cholesterol, triglyceride, low-density lipoprotein cholesterol and fasting glucose level, heterogeneity in genetic variances explained by variants with HWE was observed only for body mass index and triglyceride (P<0.05).27
Sex-specific selection against particular genetic variants might be a plausible explanation for the unequal sex ratio in humans although it is difficult to explain the 1.02–1.08 sex ratio with the current findings. This study showed female-specific selection at the DNAH11 association signal and male-specific selection at the PPP1R12B association signal, with different effect sizes. A presumable scenario is that sex ratio can be maintained by a balance of power between female-specific selection and male-specific selection, but is not exactly equal to 1:1.
In conclusion, the current study revealed two novel genetic association signals influencing sex ratio. This implied that genetic variants in proximity to the association signals might influence sex ratio. Fine mapping is needed to track down causal genetic variants responsible for sex ratio distortion. Additional studies with rare and sex-chromosomal variants would be helpful to understand the genetic architecture of sex ratio distortion. Further studies are necessary to reveal the mechanisms underlying sex ratio distortion.
References
James, W. H. The human sex ratio. Part 1: A review of the literature. Hum. Biol. 59, 721–752 (1987).
Mackenzie, C. A., Lockridge, A. & Keith, M Declining sex ratio in first nation community. Environ. Health Perspect. 113, 1295–1298 (2005).
Kaba, A. J. Sex ratio at birth and racial differences: why do black women give birth more females than non-black women? Afr. J. Reprod. Health 12, 139–150 (2008).
Chahnazarian, A. Determinants of the sex ratio at birth: review of recent literature. Soc. Biol. 35, 214–235 (1988).
Dodds, L. & Armson, B. A. Is Canada’s sex ratio in decline? CMAJ 156, 46–48 (1997).
Fukuda, M., Fukuda, K., Shimizu, T., Andersen, C. Y. & Byskov, A. G. Parental periconceptional smoking and male: female ratio of newborn infants. Lancet 359, 1407–1408 (2002).
James, W. H. Further evidence that mammalian sex ratios at birth are partially controlled by parental hormone levels around the time of conception. Hum. Reprod. 19, 1250–1256 (2004).
Trichopoulos, D. Evidence of genetic variation in the human sex ratio. Hum. Biol. 39, 170–175 (1967).
Curtsinger, J. W., Ito, R. & Hiraizumi, Y. A two-generation study of human sex-ratio variation. Am. J. Hum. Genet. 35, 951–961 (1983).
Gellatly, C Trends in population sex ratios may be explained by changes in the frequencies of polymorphic alleles of a sex ratio gene. Evol. Biol 36, 190–200 (2009).
Boraska, V., Jerončić, A., Colonna, V., Southam, L., Nyholt, D. R., Rayner, N. W. et al. Genome-wide meta-analysis of common variant differences between men and women. Hum. Mol. Genet 21, 4805–4815 (2012).
Cho, Y. S., Go, M. J., Kim, Y. J., Heo, J. Y., Oh, J. H., Ban, H. J. et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat. Genet. 41, 527–534 (2009).
Rabbee, N. & Speed, T. P. A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics 22, 7–12 (2006).
Shin, J. & Lee, C. A mixed model reduces spurious genetic associations produced by population stratification in genome-wide association studies. Genomics 105, 191–196 (2015).
Lippert, C., Listgarten, J., Liu, Y., Kadie, C. M., Davidson, R. I. & Heckerman, D. FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–835 (2011).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Kong, M., Kim, Y. & Lee, C. A strong synergistic epistasis between FAM134B and TNFRSF19 on the susceptibility to vascular dementia. Psychiatr. Genet. 21, 37–41 (2011).
Kim, Y., Kong, M. & Lee, C. Association of intronic sequence variant in the gene encoding spleen tyrosine kinase with susceptibility to vascular dementia. World J. Biol. Psychiatry 14, 220–226 (2013).
Knowles, M. R., Leigh, M. W., Carson, J. L., Davis, S. D., Dell, S. D., Ferkol, T. W. et al. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax 67, 433–441 (2012).
Leigh, M. W., Pittman, J. E., Carson, J. L., Ferkol, T. W., Dell, S. D., Davis, S. D. et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet. Med. 11, 473–487 (2009).
Visconti, P. E. & Kopf, G. S. Regulation of protein phosphorylation during sperm capacitation. Biol. Reprod. 59, 1–6 (1998).
Buffone, M. G., Doncel, G. F., Marín Briggiler, C. I., Vazquez-Levin, M. H. & Calamera, J. C. Human sperm subpopulations: relationship between functional quality and protein tyrosine phosphorylation. Hum. Reprod. 19, 139–146 (2004).
Deng, L., Zhang, D., Richards, E., Tang, X., Fang, J., Long, F. et al. Constructing an initial map of transmission distortion based on high density HapMap SNPs across the human autosomes. J. Genet. Genomics 36, 703–709 (2009).
Chakrabarti, R., Cheng, L., Puri, P., Soler, D. & Vijayaraghavan, S. Protein phosphatase PP1 gamma 2 in sperm morphogenesis and epididymal initiation of sperm motility. Asian J. Androl. 9, 445–452 (2007).
Su, A. I., Wiltshire, T., Batalov, S., Lapp, H., Ching, K. A., Block, D. et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl Acad. Sci. USA 101, 6062–6067 (2004).
Lee, D. & Lee, C. Age- and gender-dependent heterogeneous proportion of variation explained by SNPs in quantitative traits reflecting human health. Age 37, 19 (2015).
Acknowledgements
We are grateful to the National Institute of Health in Korea for providing the genotypic and epidemiological data to the KARE Analysis Consortium. This study was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (Grant No. 2012002096).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Ryu, D., Ryu, J. & Lee, C. Genome-wide association study reveals sex-specific selection signals against autosomal nucleotide variants. J Hum Genet 61, 423–426 (2016). https://doi.org/10.1038/jhg.2015.169
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.169
