
Abstract
The diagnosis of the genetic etiology of deafness contributes to the clinical management of patients. We performed the following four genetic tests in three stages for 52 consecutive deafness subjects in one facility. We used the Invader assay for 46 mutations in 13 genes and Sanger sequencing for the GJB2 gene or SLC26A4 gene in the first-stage test, the TaqMan genotyping assay in the second-stage test and targeted exon sequencing using massively parallel DNA sequencing in the third-stage test. Overall, we identified the genetic cause in 40% (21/52) of patients. The diagnostic rates of autosomal dominant, autosomal recessive and sporadic cases were 50%, 60% and 34%, respectively. When the sporadic cases with congenital and severe hearing loss were selected, the diagnostic rate rose to 48%. The combination approach using these genetic tests appears to be useful as a diagnostic tool for deafness patients. We recommended that genetic testing for the screening of common mutations in deafness genes using the Invader assay or TaqMan genotyping assay be performed as the initial evaluation. For the remaining undiagnosed cases, targeted exon sequencing using massively parallel DNA sequencing is clinically and economically beneficial.
Similar content being viewed by others

Introduction
Congenital hearing loss is a common sensory disorder, affecting approximately one per 1000 live births. Genetic factors are the most common etiology of congenital sensorineural hearing loss (SNHL), which comprises at least 50% of all cases of SNHL. Approximately 70% of cases of hereditary SNHL are non-syndromic, with the remaining 30% being syndromic.1 With regard to non-syndromic hearing loss, 75–80% of cases are inherited in an autosomal recessive (AR) pattern, 20–25% are inherited in an autosomal dominant (AD) pattern and 1–1.5% are inherited in an X-linked pattern.1 However, SNHL exhibits extremely heterogeneous disease traits. More than 140 loci have been mapped and over 80 genes have been reported to be responsible for hereditary non-syndromic hearing loss (Hereditary Hearing loss Homepage, http://hereditaryhearingloss.org/). Genetic testing has the potential to allow accurate diagnosis of SNHL, and the diagnosis of the genetic etiology contributes to the clinical management of patients, including the selection of the most appropriate treatment. For example, early therapeutic interventions for associated symptoms including diabetes mellitus or visual impairment can be performed. Further, we can predict the type of hearing loss and its progressiveness, and choose the appropriate mode of intervention from among hearing aids, electric acoustic stimulation and cochlear implantation.
In Japan, genetic testing for 46 known mutations in 13 common deafness genes using Invader assay technology became clinically available as a molecular diagnostic tool from April 2012. The cost of this genetic testing is fully covered by public health insurance, after approval from the Ministry of Health, Labor and Welfare. This genetic testing panel can analyze known single-nucleotide variants or small deletions that are commonly found in Japanese SNHL patients, as established from the results of several studies.2, 3, 4, 5 A previous study reported that the Invader assay for screening 46 known mutations in 13 deafness genes was able to identify the causative single-nucleotide variants in 28.0% (74/264) of Japanese deafness patients.6 To increase the diagnostic rate, we speculated that the addition of other genetic tests was required.
In this study, we evaluated consecutive patients who underwent genetic testing for SNHL in Yokohama City University Hospital and Yokohama City University Medical Center. To improve the diagnostic rate while giving appropriate consideration to the cost of testing, we performed the following four genetic tests in three stages: (1) the Invader assay that we applied to the screening of 46 known mutations in 13 deafness genes and Sanger sequencing for the GJB2 gene or SLC26A4 gene; (2) the TaqMan genotyping assay that we applied to the test of screening of 55 known mutations in 6 deafness genes; and (3) targeted exon sequencing using massively parallel DNA sequencing (MPS) for the testing of the sequence of 63 known deafness genes.
The goals of this study were to elucidate (1) the utility of the genetic tests performed in one facility for consecutive patients during the study period; (2) the possibility of integrating several methods of genetic testing, sequentially performed, into a routine genetic testing program; and (3) the differences in the clinical presentations of diagnosed and undiagnosed cases.
Subjects and methods
Subjects
We recruited 59 consecutive Japanese subjects with SNHL who visited two hospitals (Yokohama City University Hospital and Yokohama City University Medical Center) between July 2012 and November 2014. The patients who visited Yokohama City University Medical Center were referred to Yokohama City University Hospital for genetic testing. Basically, all subjects presenting with bilateral SNHL were included. The exclusion criteria were cases with chromosomal abnormalities, such as a 21 trisomy, and cases with SNHL caused by middle ear disease or ear surgery. All subjects were unrelated and from non-consanguineous families. For each proband and their family members, informed written consent was obtained to participate in this study, which was approved by the Human Subjects Ethical Committee of Yokohama City University.
Among the 59 recruited cases, 5 cases of 21 trisomy and 1 case of Kabuki make-up syndrome were excluded from this study. One additional case refused to undergo genetic testing, so that 52 cases were enrolled in this study. Twenty patients were male and 32 were female. Fifty-one patients were Japanese, and one was half Filipino and half Japanese. The age at the time of enrollment ranged from 0 to 76 years (median: 4 years). The age of onset was between 0 and 66 years (median: 0). As shown in Table 1, 32 cases (62%) had congenital SNHL, with hearing loss detected by newborn hearing screening at birth in 27 cases. The other five cases were referred to our hospital with suspected hearing loss, even though newborn hearing screening was not performed, and were diagnosed with congenital SNHL based on their general condition. With regard to the mode of inheritance, 12 cases were AD inheritance, 5 were AR inheritance and 35 were sporadic cases. Forty-four cases had symmetric hearing loss, and eight cases had asymmetric SNHL with a >10 dB difference in right–left laterality.
Four of the 54 patients presented with various physical complications. One case had retinitis pigmentosa associated with Usher syndrome, one case had decolorization of the iris pigment associated with Waardenburg syndrome, one case had palmoplantar keratoderma associated with syndromic SNHL caused by GJB2 mutations and one case had bilateral aural fistulas associated with branchio-oto-renal syndrome (BOR syndrome).
Clinical information and blood samples were obtained from each proband and from all consenting affected and unaffected relatives. The following data were collected. (1) Pure-tone audiograms, behavioral audiometry or auditory brainstem responses. (2) Medical history including the age of onset, defined as when patients noticed or were diagnosed with hearing loss, and complications. Inheritance was classified as AD, hearing loss passed down through families; AR, affecting siblings; or sporadic, no family history of hearing loss. (3) Temporal bone imaging (computed tomography and/or magnetic resonance) was undertaken where applicable.
Age of onset was divided into three categories: congenital, SNHL was detected by newborn hearing loss screening or was strongly suspected to be present at birth; childhood, SNHL was diagnosed before 17 years of age and was not categorized as congenital; and adult, SNHL was diagnosed after 18 years of age. Pure-tone audiograms and/or auditory brainstem responses were used to confirm the degree and progression of SNHL. Hearing levels were classified based on the better hearing ear as: normal, <20 dB; mild hearing loss, 21–40 dB; moderate hearing loss, 41–70 dB; severe hearing loss, 71–95 dB; and profound hearing loss, >95 dB. Differences in hearing thresholds of less than 10 dB are commonly observed as a result of imprecisions in audiometry.7 We, therefore, defined asymmetric SNHL as an average difference of over 10 dB in hearing thresholds between ears (measured at 0.5, 1, 2 and 4 kHz).
Methods
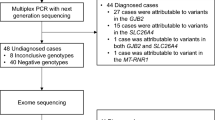
As shown in Figure 1, this study consisted of a three-stage genetic testing program.

Flow diagram of the genetic testing. Four types of genetic testing were performed in three stages. (+) Possible variant(s) identified, (–) no variant(s) identified, *Massively parallel DNA sequencing.
First-stage testing: the Invader assay and Sanger sequencing for GJB2 and SLC26A4
The Invader assay was applied to all patients enrolled in this study for the screening of 46 known mutations in 13 known deafness genes. The detailed protocol was described elsewhere.6 In brief, 1.2 µl of primary probe/Invader oligonucleotide mixture (containing 0.5 µmol l–1 wild-type primary probe, 0.5 µmol l–1 mutant primary probe, 0.05 µmol l–1 Invader oligonucleotide and 10 mmol l–1 3-(N-morpholino)propanesulfonic acid (MOPS)) were added to each well of 384-well plates. Fluorescent resonance energy transfer/cleavase mixture (Third Wave Technologies, Madison, WI, USA) was added to the plates containing the probe/Invader oligonucleotide mixture. Next, 3 µl of 5–100 fmol l–1 synthetic target oligonucleotides (positive control), 10 µg ml–1 yeast tRNA (no target control) and denatured genomic DNA samples (0.15 ng µl–1) were added. A measure of 6 µl of mineral oil (Sigma, St Louis, MO, USA) was laid over all the reaction wells, which were then incubated at 63 °C for 4 h. Incubation fluorescence was then measured using a CytoFluor 4000 fluorescent microplate reader (Thermo Fisher Scientific, Waltham, MA USA). Mutations commonly found in Japanese were then selected (GJB2 (12 mutations), SLC26A4 (19 mutations), COCH (one mutation), KCNQ4 (one mutation), MYO7A (one mutation), TECTA (two mutations), CRYM (two mutations), POU3F4 (one mutation), EYA1 (three mutations), mitochondrial 12 S ribosomal RNA, mitochondrial tRNA (Leu), mitochondrial tRNA (Ser) and mitochondrial tRNA (Lys)). All mutations are shown in Supplementary Table 1.
After performing the Invader assay, Sanger sequencing for GJB2 or SLC26A4 containing the entire coding region was performed for heterozygous cases, in which one allelic mutation was found in the GJB2 or SLC26A4 gene.
Second-stage testing: the TaqMan genotyping assay
For cases in which no causative mutations were identified in the first-stage genetic testing, the TaqMan SNP genotyping assay was applied (Thermo Fisher Scientific). The detailed protocol was described elsewhere.8 We selected probes for 55 known mutations in 6 deafness genes (SLC26A4 (15 mutations), CDH23 (22 mutations), KCNQ4 (one mutation), TMPRSS3 (5 mutations), OTOF (9 mutations) and MYO15A (3 mutations)). All mutations are shown in Supplementary Table 2.
Third-stage testing: targeted exon sequencing using MPS
For cases in which no causative mutations were identified in the first- and second-stage testing, targeted exon sequencing using MPS was applied.
Amplicon library preparation
Amplicon libraries were prepared using an Ion AmpliSeq Custom Panel (Thermo Fisher Scientific) for 63 genes reported to cause non-syndromic hearing loss according to the manufacturer’s instructions. All mutations are shown in Supplementary Table 3. The detailed protocol was described elsewhere.9 After preparation, the amplicon libraries were diluted to 20 pm and equal amounts of six libraries for six patients were pooled for one sequence reaction.
Emulsion PCR and sequencing
Emulsion PCR and sequencing were performed according to the manufacturer’s instructions. The detailed protocol was described elsewhere.9 MPS was performed with an Ion Torrent Personal Genome Machine (PGM) system using an Ion PGM 200 Sequencing Kit and an Ion 318 Chip (Thermo Fisher Scientific).
Base call and data analysis
The sequence data were mapped against the human genome sequence (build GRCh37/hg19) with a Torrent Mapping Alignment Program. After sequence mapping, the DNA variant regions were piled up with Torrent Variant Caller plug-in software. After variant detection, their effects were analyzed using ANNOVAR software.10, 11 The missense, nonsense, insertion/deletion and splicing variants were selected from among the identified variants. Variants were further selected as less than 1% of (1) the 1000 genome database (http://www.1000genomes.org/), (2) the 6500 exome variants (http://evs.gs.washington.edu/EVS/), (3) the Human Genetic Variation Database (data set for 1208 Japanese exome variants; http://www.genome.med.kyoto-u.ac.jp/SnpDB/index.html) and (4) the 269 in-house Japanese normal hearing controls. To predict the pathogenicity of missense variants, the following functional prediction softwares were used: PhyloP (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/phyloP44way/), sorting intolerant from tolerant (SIFT; http://sift.jcvi.org/), polymorphism phenotyping (PolyPhen2; http://genetics.bwh.harvard.edu/pph2/), LRT (http://www.genetics.wustl.edu/jflab/lrt_query.html), MutationTaster (http://www.mutationtaster.org/) and GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html). The sequencing data were available in the DDBJ databank of Japan (Accession number: JGAS00000000032).
Variant confirmation
Candidate mutations were confirmed by Sanger sequencing, and the responsible mutations were identified by segregation analysis using samples from among the patients’ family members.
Results
Diagnostic rate of genetic testing
As shown in Figure 2 and Table 1, we performed genetic testing using the Invader assay for all 52 patients. Five patients were identified as having biallelic homozygous or compound heterozygous mutations in GJB2 (10%, 5/52). A further six patients were identified with heterozygous GJB2 or SLC26A4 mutations in one allele. We performed Sanger sequencing for the entire GJB2 or SLC26A4 gene in these six cases. On the basis of the results of the Sanger sequencing, four cases were diagnosed as having biallelic compound heterozygous mutations in GJB2 or SLC26A4, and the remaining two cases were diagnosed as heterozygous carriers of GJB2 or SLC26A4 mutations. Finally, seven patients were diagnosed with SNHL caused by GJB2 mutations. Two patients had SNHL caused by SLC26A4 mutations, as shown in Table 2, with both cases showing bilateral enlargement of the vestibular aqueduct. The detection and diagnostic rates of the first-stage testing were 21% (11/52) and 17% (9/52), respectively.

Results of genetic testing. Five cases were diagnosed by the Invader assay. Four cases were diagnosed by the Invader assay and direct sequencing for GJB2 or SLC26A4. Four cases were diagnosed by second-stage testing using the TaqMan genotyping assay. Eight cases were diagnosed by targeted exon sequencing using the massively parallel DNA sequencing. *Massively parallel DNA sequencing.
We next performed second-stage testing using the TaqMan genotyping assay for the 43 patients who could not be diagnosed by the first-stage testing. Three patients with AD inheritance were identified as having heterozygous KCNQ4 mutations. One case with AR inheritance was identified as having biallelic compound heterozygous mutations in CDH23. As shown in Table 3, two cases were also found to be heterozygous carriers of CDH23 mutations. In addition, one patient was a heterozygous carrier of OTOF mutations. The detection and diagnostic rates of the second-stage testing were 16% (7/43) and 9% (4/43), respectively.
We performed third-stage testing with targeted exon sequencing using MPS for the 39 patients who could not be diagnosed by the first- and second-stage testing. Twenty-two variants of 11 genes, including 10 novel mutations, were identified in 16 cases (Table 3). A combination of Sanger sequencing and family segregation analysis confirmed that six cases had biallelic causative homozygous or compound heterozygous mutations in the PTPRQ, LOXHD1, CDH23, MYO15A, USH2A and SLC26A4 genes, and two cases with AD inheritance had causative heterozygous mutations in the ACTG1 and WFS1 genes, respectively (Figure 2 and Table 2). Twenty-two variants were diagnosed as pathologic for the following reasons. (1) Twelve variants have already been reported as pathologic mutations (SLC26A4; c.1229C>T20, c.1625C>G21, c.1489G>A22, c.1707+5G>A23: CDH23; c.6085C>T18, c.4762C>T18, c.719C>T18: OTOF; c.1194T>A24: WFS1; c.2508G>T19: MYO7A; c.652G>A25: TMPRSS3; c.778G>A9: MYO15A; c.6371G>A26). (2) Among 10 variants, 9 variants (CDH23; c.6555G>A, PTPRQ; c.1261C>T: MYO15A; c.3756+1G>A, c.4660G>A: LOXHD1; c.G3076G>T, c.4375+1G>T: USH2A; c.57_58del, c.9079G>T: ACTG1; c.994C>T) were not found in the 1000 genome database, the 6500 exome variants or the Human Genetic Variation Database, suggesting those variants are pathologic. One variant (CDH23; c.1363G>A) was found in 0.16% of the 6500 exome variants but not in the 1000 genome database or the Human Genetic Variation Database, which is also compatible with it being a pathologic mutation. Segregation analysis also confirmed their association with the affected family members. (3) Out of the 10 novel mutations, 5 were nonsense mutations, splice site mutations, or frameshift mutations, indicating that they are likely to be pathologic. (4) Four novel missense mutations were predicted as pathogenic by PhyloP, SIFT, PolyPhen2, LRT, MutationTaster and GERP++. (5) One novel missense mutation (ACTG1, c.994C>T, p.322S) was reported in which the amino acid was located near the adenine-binding site and is speculated to be important for protein function.27 Further, this amino acid is conserved among vertebrates and invertebrates.28
The case with compound heterozygous mutations in SLC26A4 also showed bilateral enlargement of the vestibular aqueduct. The detection and diagnostic rates of third-stage testing were 41% (16/39) and 21% (8/39), respectively.
Diagnostic rate and clinical presentation
As shown in Table 1, the diagnostic rates of AD, AR and sporadic cases were 50% (6/12), 60% (3/5) and 34% (12/35), respectively. When classified by age of onset, our genetic testing resolved 43% (14/32), 31% (5/16) and 50% (2/4) of congenital, childhood- and adult-onset cases, respectively. The diagnostic rate divided by the degree of hearing level was 29% (6/21) and 48% (15/31) for mild-to-moderate and severe-to-profound SNHL, respectively. Our genetic testing resolved 45% (20/44) of the cases with symmetric SNHL, whereas there was only one case that was diagnosed with genetic SNHL among the cases with asymmetric SNHL (1/8).
As shown in Table 4, the diagnostic rates of sporadic cases with congenital, childhood- and adult-onset SNHL were 42% (11/26), 13% (1/8) and 0% (0/1), respectively. Among sporadic cases with congenital SNHL, the diagnostic rate of cases with severe-to-profound SNHL rose to 48% (10/21). Among cases with mild-to-moderate with SNHL, 44% (4/9) of AD cases were diagnosed. In contrast, the diagnostic rates of AR and sporadic cases with mild-to-moderate SNHL were 0% (0/1) and 18% (2/11), respectively.
Discussion
Yokohama, which abuts the western edge of Tokyo, is the capital of Kanagawa prefecture and the second largest city in Japan. The population of Kanagawa prefecture and Yokohama is ~9 million and 3.7 million, respectively, and population turnover rates are high. Our two hospitals, coordinated by the Yokohama City University School of Medicine, are located in the center of Yokohama, and all enrolled cases came from the Yokohama area. The levels of consanguinity and population genetic bias are thought to be low. Also, this study was designed to minimize sampling and selection bias.
The diagnostic rates of the first-, second- and third-stage genetic testing were 17%, 9% and 21%, respectively (Table 1). Genetic diagnosis was possible in 13 cases using the first- and second-stage testing (Figure 1). In the third stage, novel mutations were also identified in rare deafness genes such as PTPRQ or LOXHD1 (Table 2), and we were able to diagnose eight cases with genetic SNHL (Figure 1).
The Invader assay enables the screening for 13 common genes and 46 common single-nucleotide variants, small insertion or deletions. To increase the number of common pathogenic variants, we performed the TaqMan genotyping assay, which includes 55 mutations in six genes previously reported to cause deafness. With respect to the results of the screening of common deafness variants using the Invader assay and TaqMan typing assay, 25% (13/52) of cases could be diagnosed. Our results suggest that the majority of the cases can be identified by genetic testing with common variant screening. These results are consistent with those of our recent genetic epidemiological study29, 30.
Targeted exon sequencing using MPS is a method for the simultaneous sequencing of large numbers of known genes31, 32 and has recently been applied to clinical diagnostic testing. Targeted exon sequencing using MPS has the greater advantage of making it possible to identify rare or novel variants in deafness genes. The common mutations included in the Invader assay panel and TaqMan probes would also be identified by targeted exon sequencing using MPS. However, until recently there have been a number of limitations; for example (1) mitochondrial DNA, which has many copies, is not suitable for MPS in terms of cost effectiveness and (2) particular variants, especially those located in the GC-rich region, could not be identified.
The Invader assay, for example, is an effective method for identifying mitochondrial mutations and their heteroplasmy rates. Therefore, the Invader assay is still used in combination with the MPS-based genetic testing, which began to be applied from August 2015 as a social health insurance-based screening method in Japan. Further, TaqMan genotyping is very effective in identifying particular mutations (for example, c.211delC in KCNQ4), which cannot be detected by MPS-based screening.
For a comprehensive screening system, it is therefore necessary to combine the Invader assay, TaqMan genotyping assay and targeted exon sequencing using MPS technology.
As shown in Table 1, the diagnostic rate of AD was 50% (6/12) and AR cases was 60% (3/5), whereas the diagnostic rate of sporadic cases fell to 34% (12/35). However, as shown in Table 4, the diagnostic rate of sporadic cases with congenital and severe-to-profound SNHL was 48% (10/21). Family history is important for genetic diagnosis. In most developed countries, many AR cases are recognized as sporadic cases as the size of the family is falling due to the low birth rate. Genetic testing is valuable, even for sporadic cases, when congenital and severe SNHL is suspected.
The diagnostic rates of sporadic cases with mild-to-moderate SNHL or with childhood- and adult-onset SNHL were lower than those of other cases (Table 4). The diagnostic rate of non-dominant cases with mild-to-moderate SNHL was previously reported to be lower than the diagnostic rate of non-dominant cases with severe-to-profound SNHL.33 One possible reason for this is that we did not analyze copy number variations, which is a common cause of mild-to-moderate SNHL.31, 34 Late-onset SNHL and asymmetric SNHL are attributable to environmental or multiple genetic effects rather than to single genetic effects.35 We identified a variant case that had compound heterozygous mutations in SLC26A4 among eight cases with asymmetric SNHL. The average difference in hearing threshold between ears was 13 dB, and laterality was only detected at low frequencies. Computed tomography images revealed enlargement of the vestibular aqueduct in both inner ears. Shearer et al. also reported the diagnostic rate for cases with asymmetric SNHL to be 0%.31 It is difficult to recommend genetic testing for sporadic cases with childhood- or adult-onset SNHL or for cases with asymmetric SNHL. However, we will consider applying genetic testing in accordance with the clinical details of each individual.
In conclusion, we identified the genetic cause of SNHL in 40% (21/52) of cases using the unified three-stage genetic testing. The combined approach involving the screening of known mutations commonly found in the Japanese population with the Invader and TaqMan genotyping assays, and targeted exon sequencing using MPS appears to be a useful diagnostic tool, and there appear to be significant benefits to the incorporation of these technologies as part of a routine diagnostic program.
References
Van Camp, G., Willems, P. & Smith, R. J. Nonsyndromic hearing impairment: unparalleled heterogeneity. Am. J. Hum. Genet. 60, 758–764 (1997).
Ohtsuka, A., Yuge, I., Kimura, S., Namba, A., Abe, S., Van Laer, L. et al. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum. Genet. 112, 329–333 (2003).
Oguchi, T., Ohtsuka, A., Hashimoto, S., Oshima, A., Abe, S., Kobayashi, Y. et al. Clinical features of patients with GJB2 (connexin 26) mutations: severity of hearing loss is correlated with genotypes and protein expression patterns. J. Hum. Genet. 50, 76–83 (2005).
Suzuki, H., Oshima, A., Tsukamoto, K., Abe, S., Kumakawa, K., Nagai, K. et al. Clinical characteristics and genotype-phenotype correlation of hearing loss patients with SLC26A4 mutations. Acta Otolaryngol. 127, 1292–1297 (2007).
Usami, S. I., Wagatsuma, M., Fukuoka, H., Suzuki, H., Tsukada, K., Nishio, S. Y. et al. The responsible genes in Japanese deafness patients and clinical application using Invader assay. Acta Otolaryngol. 128, 446–454 (2008).
Usami, S. I., Nishio, S. Y., Nagano, M., Abe, S. & Yamaguchi, T. Simultaneous screening of multiple mutations by invader assay improves molecular diagnosis of hereditary hearing loss: a multicenter study. PLos ONE 7, e31276 (2012).
Lutman, M. E. & Coles, R. R. A. Asymmetric sensorineural hearing thresholds in the non-noise-exposed UK population: a retrospective analysis. Clin. Otolaryngol. 34, 316–321 (2009).
De Kok, J. B., Wiegerinck, E. T., Giesendorf, B. A. & Swinkels, D. W. Rapid genotyping of single nucleotide polymorphisms using novel minor groove binding DNA oligonucleotides (MGB probes). Hum. Mutat. 19, 554–559 (2002).
Miyagawa, M., Nishio, S. Y., Ikeda, T., Fukushima, K. & Usami, S. Massively parallel DNA sequencing successfully identifies new causative mutations in deafness genes in patients with cochlear implantation and EAS. PLoS ONE 8, e75793 (2013).
Chang, X. & Wang, K. wANNOVAR; annotating genetic variants for personal genomes via the web. J. Med. Genet. 49, 433–436 (2012).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variation from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Fuse, Y., Doi, K., Hasegawa, T., Squii, A., Hibino, H. & Kudo, T. Three novel connexin26 gene mutations in autosomal recessive non-syndromic deafness. Neuroreport 10, 1853–1857 (1999).
Richard, G., White, T. W., Smith, L. E., Bailey, R. A., Compton, J. G., Paul, D. L. et al. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum. Genet. 103, 393–339 (1998).
Yuge, I., Ohtsuka, A., Matsunaga, T. & Usami, S. Identification of 605ins46, a novel GJB2 mutation in a Japanese family. Auris Nasus Larynx 29, 379–382 (2002).
Van, Hauwe, P., Everett, L. A., Coucke, P., Scotto, D. A., Kraft, M. L., Ris-Stalpers, C. et al. Two frequent missense mutations in Pendred syndrome. Hum. Mol. Genet. 7, 1099–1104 (1998).
Naito, T., Nishio, S. Y., Iwasa, Y., Yano, T., Kumakawa, K., Abe, S. et al. Comprehensive genetic screening of KCNQ4 in a large autosomal dominant nonsyndromic hearing loss cohort: genotype-phenotype correlations and a founder mutation. PLoS One 8, e63231 (2013).
Wagatsuma, M., Kitoh, R., Suzuki, H., Fukuoka, H., Takumi, Y. & Usami, S. Distribution and frequencies of CDH23 mutations in Japanese patients with non-syndromic hearing loss. Clin. Genet. 72, 339–344 (2007).
Miyagawa, M., Nishio, SY. & Usami, S. Prevalence and clinical features of hearing loss patients with CDH23 mutations: a large cohort study. PLoS ONE 7, e40366 (2012).
Hogewind, B. F., Pennings, R. J., Hol, F. A., Kunst, H. P., Hoefsloot, E. H., Cruysberg, J. R. et al. Autosomal dominant optic neuropathy and sensorineual hearing loss associated with a novel mutation of WFS1. Mol. Vis. 16, 26–35 (2010).
Miyagawa, M., Nishio, S. Y. & Usami, S. Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: a large cohort study. J. Hum. Genet. 59, 262–268 (2014).
Choi, B. Y., Stewart, A. K., Nishimura, K. K., Cha, W. J., Seong, M. W., Park, S. S. et al. Efficient molecular genetic diagnosis of enlarged vestibular aqueducts in East Asians. Genet. Test. Mol. Biomarkers 13, 679–687 (2009).
Li, X. C., Everett, L. A., Lalwani, A. K., Desmukh, D., Fridman, T. B., Green, E. D. et al. A mutation in PDS causes non-syndromic recessive deafness. Nat. Genet. 18, 215–217 (1998).
Park, H. J., Lee, S. J., Jin, H. S., Lee, J. O., Go, S. H., Jong, H. S. et al. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin. Genet. 67, 160–165 (2005).
Iwasa, Y., Nishio, S. Y., Yoshimura, H., Kanda, Y., Kumakawa, K., Abe, S. et al. OTOF mutation screening in Japanese severe to profound recessive hearing loss patients. BMC Med. Genet. 14, 95 (2013).
Sun, Y., Chen, J., Sun, H., Cheng, J., Li, J., Lu, Y. et al. Novel missense mutations in MYO7A underlying postlingual high- or low-frequency non-syndromic hearing impairment in two large families from China. J. Hum. Genet. 56, 64–70 (2011).
Shearer, A. E., Hildebrand, M. S., Webster, J. A., Kahrizi, K., Meyer, N. C., Jalavand, K. et al. Mutations in the first MyTH4 domain of MYO15A are a common cause of DFNB3 hearing loss. Laryngoscope 119, 727–733 (2009).
Bryan, K. E, Wen, K. K, Zhu, M, Rendtorff, N. D, Feldkamp, M, Tranebjaerg, L et al. Effects of human deafness γ-actin mutations (DFNA20/26) on actin function. J. Biol. Chem. 281, 20129–20139 (2006).
Zhu, M., Yang, T., Wei, S., DeWan, A. T., Morell, R. J. & Elfenbein, J. L. Mutations in the g-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am. J. Hum. Genet. 73, 1082–1091 (2003).
Miyagawa, M., Naito, T., Nishio, S. Y., Kamatani, N. & Usami, S. Targeted exon sequencing successfully discovers rare causative genes and clarifies the molecular epidemiology of Japanese deafness patients. PLoS ONE 8, e71381 (2013).
Nishio, S. Y. & Usami, S. I. Deafness gene variations in a 1120 nonsyndromic hearing loss cohort molecular epidemiology and deafness mutation spectrum of patients in Japan. Ann. Otol. Rhinol. Laryngol. 124 (Suppl 1), 49S–60S (2015).
Shearer, A. E., Black-Ziegelbein, E. A., Hildebrand, M. S., Eppsteiner, R. W., Ravi, H. et al. Advancing genetic testing for deafness with genomic technology. J. Med. Genet. 50, 627–634 (2013).
Shearer, A. E., DeLuca, A. P., Hildebrand, M. S., Taylorc, K. R., Gurrola, J. II, Scherer, S. et al. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc. Natl Acad. Sci. USA 107, 21104–21109 (2010).
Chai, Y., Pang, X., Chen, D., Li, L., Chen, Y., Sun, L. et al. Molecular etiology of non-dominant, non-syndromic, mild-to-moderate childhood hearing impairment in Chinese Hans. Am. J. Med. Genet. A 164A, 3115–3119 (2014).
Shearer, A. E., Kolbe, D. L., Azaiez, H., Sloan, C. M., Frees, K. L., Weaver, A. E. et al. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 22, 37 (2014).
Kochhar, A., Hildebrand, M. S. & Smith, R. J. Clinical aspects of hereditary hearing loss. Genet. Med. 9, 393–408 (2007).
Acknowledgements
This study was supported by a Health and Labour Sciences Research Grant for Research on Rare and Intractable Diseases and Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labour and Welfare of Japan (SU), by Practical Research Project for Rare / Intractable Disease from Japan Agency for Medical Research and development (AMED), and by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture of Japan (SU). We thank Dr Yasunori Sakuma for his help about collecting the clinical information.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Sakuma, N., Moteki, H., Takahashi, M. et al. An effective screening strategy for deafness in combination with a next-generation sequencing platform: a consecutive analysis. J Hum Genet 61, 253–261 (2016). https://doi.org/10.1038/jhg.2015.143
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.143
This article is cited by
-
Addition of an affected family member to a previously ascertained autosomal recessive nonsyndromic hearing loss pedigree and systematic phenotype-genotype analysis of splice-site variants in MYO15A
BMC Medical Genomics (2022)
-
Exome sequencing in infants with congenital hearing impairment: a population-based cohort study
European Journal of Human Genetics (2020)
-
Whole-exome sequencing identifies a novel missense variant within LOXHD1 causing rare hearing loss in a Chinese family
BMC Medical Genetics (2019)
-
Novel Usher syndrome pathogenic variants identified in cases with hearing and vision loss
BMC Medical Genetics (2019)
-
Utilization of amplicon-based targeted sequencing panel for the massively parallel sequencing of sporadic hearing impairment patients from Saudi Arabia
BMC Medical Genetics (2016)
