
Abstract
Spinal muscular atrophy (SMA) is caused by functional loss of the survival of motor neuron 1 (SMN1) gene. Despite genetic homogeneity, phenotypic variability indicates the involvement of disease modifiers. SMN1 is located in 5q13.2 segmental duplication, enriched in genes and prone to unequal rearrangements, which results in copy number polymorphism (CNP). We examined the influence of CNP of 5q13.2 genes and their joint effect on childhood-onset SMA phenotype. Multiplex ligation-dependent probe amplification (MLPA) was used to construct 5q13.2 alleles and assess copy number of the SMN2, small EDRK-rich factor 1A (SERF1A) and NLR family apoptosis inhibitory protein (NAIP) genes in 99 Serbian patients with SMN1 homozygous absence (23-type I, 37-type II and 39-mild type III) and 122 patients' parents. Spearman rank test was performed to test correlation of individual genes and SMA type. Generalized linear models and backward selection were performed to obtain a model explaining phenotypic variation with the smallest set of variables. 5q13.2 alleles most commonly associated with type I harbored large-scale deletions, while those detected in types II and III originated from conversion of SMN1 to SMN2. Inverse correlation was observed between SMN2, SERF1A and NAIP CNP and SMA type (P=2.2e−16, P=4.264e−10, P=2.722e−8, respectively). The best minimal model describing phenotypic variability included SMN2 (P<2e−16), SERF1A (P<2e−16) and their interaction (P=0.02628). SMN2 and SERF1A have a joint modifying effect on childhood-onset SMA phenotype.
Similar content being viewed by others

Introduction
Spinal muscular atrophy (SMA) represents the most frequent monogenic cause of infant mortality, with an incidence of 1/6000 live births1 and a carrier rate as high as 1/35 in the European population.2 This autosomal recessive neuromuscular disorder is caused by deficiency of the survival of motor neuron (SMN) protein as a result of homozygous absence of the survival of motor neuron 1 (SMN1) gene in 96% of patients.3 Remaining patients are compound heterozygotes with an absence of one SMN1 allele and a small mutation in the other.4
Clinical presentation of SMA is extremely variable both in age at onset and severity, ranging from a severe, infantile form to a mild, adult-onset form.5, 6 Childhood-onset forms include three clinical types. Type I SMA (OMIM #253300) commences within the first 6 months after birth, patients are unable to sit unsupported and die within the first 2 years. Patients with type II SMA (OMIM #253550) are able to sit independently, but cannot walk, while patients with type III SMA (OMIM #253400), despite symmetrical proximal muscular weaknesses, gain the ability to walk and have a normal lifespan with the possibility to lose ambulation over time.
Extensive phenotypic variability and homogeneity of the disease-causing mutation clearly indicate the existence of genetic, epigenetic and environmental modifiers of the SMA clinical outcome.7 Many aspects of SMA genotype–phenotype correlation are related to the organization and rearrangements of 5q13.2 chromosomal region harboring the SMN1 gene. This region underwent segmental duplication before the separation of human and chimpanzee lineages.8 It is enriched in genes, repeated sequences and pseudogenes, which makes it highly unstable and prone to unequal rearrangements.9 Non-allelic homologous recombination causes large-scale deletions and duplications, while gene conversion increases allelic diversity in converted copies.10 Such extensive structural rearrangements result in copy number polymorphism (CNP) of 5q13.2 residing genes.11
The SMN1 gene is located in the telomeric part of the 5q13.2 segmental duplication, whereas 99% identical SMN2 gene resides in its centromeric part.3 Consequently, the absence of the SMN1 gene can be caused either by a deletion or by conversion to the SMN2 gene.12 SMN1 and SMN2 have an identical coding sequence, and the only functional difference is the synonymous substitution within exon 7 (c.840C>T), causing skipping of this exon in ~90% of SMN2 transcripts and giving rise to the non-functional SMN protein.13 The potential of the SMN2 gene to produce ~10% of the functional protein makes this gene a major genetic modifier of SMA phenotype,7 with increased copy number being associated with milder phenotype.12, 14, 15
Nevertheless, phenotypic discrepancies have been observed among patients with equal SMN2 copy number. Although some SMA genetic modifiers outside 5q13.2 region were identified,7, 16 other genes residing in this region may be involved, as they also undergo structural rearrangements. Small EDRK-rich factor 1 (SERF1 or H4F5) gene exists in two identical copies—telomeric (SERF1A) and centromeric (SERF1B), and represents the human ortholog of Caenorhabditis elegans MOAG-4, which is a general regulator of protein aggregation and proteotoxicity.17 NLR family apoptosis inhibitory protein (NAIP or BIRC1) gene exists as a full-length telomeric copy and additional pseudogene copies. The NAIP protein acts as a negative regulator of motor-neuron apoptosis.18 Previous studies showed inconsistent results regarding the correlation between SERF1A and NAIP copy number and SMA phenotype.19, 20, 21, 22, 23, 24, 25
To further clarify this issue, we determined the structure of 5q13.2 alleles in a group of Serbian childhood-onset SMA patients and their parents, analyzed the individual correlation of SMN2, SERF1A and NAIP copy number with type of childhood-onset SMA, and assessed their joint effect on SMA phenotypic outcome.
Materials and methods
Subjects
The study included 99 unrelated Serbian patients (51 females and 48 males) fulfilling clinical diagnostic criteria for SMA26 and having homozygous absence of the SMN1 gene confirmed by single-strand conformation polymorphism (SSCP) analysis3 or bidirectional SMN1 exon 7 sequencing. Patients were recruited in the period from 2001 to 2013 from two main clinics for pediatric neurology in Serbia: Clinic for Neurology and Psychiatry for Children and Youth, Belgrade and Department for Neurology, University Children's Hospital, Belgrade. To eliminate sampling bias and subjectiveness regarding the age of onset of symptoms, the ability to sit/walk unaided was taken as the main criterion in the patient classification: 23 were classified as type I, 37 as type II and 39 as type III.
The study also enrolled 122 patients’ parents willing to participate. For 56 patients, a family trio (proband and both parents) was analyzed. In 10 cases one parent was available, whereas in 33 cases none of the parents could be analyzed. Written informed consent was obtained from parents of all patients. The study was approved by Ethics Committees of the referring clinics.
Genetic analyses
Genomic DNA was extracted from blood samples using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) and quantified with the Qubit dsDNA BR Assay Kit on Qubit® 2.0 Fluorometer (Life Technologies, Grand Island, NY, USA).
Multiplex ligation-dependent probe amplification (MLPA), using the commercially available P021 A2 probe mix (MRC-Holland, Amsterdam, The Netherlands), was performed to determine the structure of the 5q13.2 region and to evaluate the copy number of the following genes: SMN1, SMN2, SERF1A, NAIP and general transcription factor II H, polypeptide 2 (GTF2H2 or p44). Approximately 200 ng of DNA was used for each MLPA reaction. Since MLPA is a relative quantification technique, all tested samples were compared with 15 reference samples using the Coffalyser software (MRC-Holland). Ratios of MLPA probes specific for the 5q13.2 genes and the assigned copy number are shown in Supplementary Table 1. A probe for the SERF1 genes in the P021 A2 probe mix is referred to as SERF1B-up, but it aligns 9167–9101 base pairs upstream from both the SERF1A and SERF1B genes (Ensembl transcripts SERF1A-002 ENST00000354833 and SERF1B-001 ENST00000380750). Therefore, after the construction of the 5q13.2 region in all examined individuals, we were able to unambiguously determine SERF1A copy number. We disregarded results of probes targeting the GTF2H2 gene due to a high CNP variability among the reference and analyzed samples, which is in accordance with previously obtained data.23 Reproducibility of the MLPA method was determined by running ~30% of randomly selected samples in independent duplicates.
In rare cases of putative de novo rearrangements, where the observed structure of the 5q13.2 region in a patient was discordant to that seen in a parent, paternity/maternity was confirmed using the AmpFlSTR Identifiler Plus PCR Amplification Kit (Life Technologies).
Statistical analyses
Individual correlation between SMN2, SERF1A and NAIP copy number and type of childhood-onset SMA, as a measure of the disease severity, was performed by Spearman rank test. To obtain the model explaining the phenotypic variation of childhood-onset SMA with the smallest set of predictor variables, the best minimal model was fitted by generalized linear models with Poisson response and log linking function. In brief, we started with a full model including the next predictor variables: three individually significantly correlated variables with childhood-onset SMA (individual copy number of the SMN2, SERF1A and NAIP genes) as well as interactions between these factors. The backward selection was performed with predictor variables being eliminated from the model in an iterative process if the significance level was >0.05. We used the Akaike information criterion for model comparison and selection, and the chosen best fitting model was the one with the lowest Akaike information criterion value. The significance level was set at 0.05 in all analyses. All statistical tests were done in R package, ver. 3.1.0.27
Results
Status of the SMN1 gene in analyzed subjects
MLPA analysis confirmed SMN1 homozygous absence in all analyzed patients. Homozygous absence encompassing the entire SMN1 gene was found in ~95% (94/99) of patients, whereas in ~5% (5/99) one SMN1 allele retained exon 8 (Figure 1).

Twenty different SERF1A/SMN1/NAIP alleles detected in 99 childhood-onset SMA patients and 122 parents. Thirteen alleles associated with the disease, found in patients and carriers, were grouped into six types (A–F) depending on the size of a rearrangement estimated through the involvement of the SERF1A and NAIP genes. Additionally, the difference between allele types A–D and types E and F is whether the SMN1 gene was completely (A–D) or partially absent (types E and F). Each of allele types A–F was further subdivided based on whether the absence of SMN1 gene was due to deletion (subtype 1) or conversion to SMN2 (subtype 2). In the E1 allele (SMN1ex8) only SMN1 exon 8 is retained, and the rest of the gene is deleted, while in E2 the rest of the gene is converted (SMNC/T). In the F1 allele, only SMN1 exon 7 is deleted (SMN1del ex7), while in SMNT/C/T only SMN1 exon 7 is converted to SMN2. Alleles G–M were found only in the parents' group. Type G carried two copies of the SMN1, and types H–L carried 0, 1, 2, 3 and 4 copies of the NAIP, respectively. Allele M carried two SMN1 and three NAIP copies. Black boxes represent a deletion, shaded boxes a gene conversion and white boxes the presence of the gene/s. Numbers of patients and parents in which the corresponding allele was observed are separated by a slash (i.e., patients/parents). If no subject was found to carry the corresponding chromosome type, the ‘−’ sign was used. The figure does not illustrate authentic physical distances between the analyzed genes nor their actual sizes.
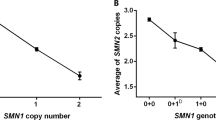
In the parents' group, carrier status was confirmed in almost all cases: 95.9% (117/122) carried one SMN1 copy (1/0 genotype or trans configuration) and 2.46% (3/122) turned out to have two SMN1 copies on the same chromosome (2/0 genotype or cis configuration). For the remaining two parents (1.64%) carrier status was not ascertained in blood cells. Positive paternity/maternity test pointed toward a de novo mutation event (one deletion and one gene conversion) or germline mosaicism.
Rearrangements and structure of the 5q13.2 telomeric region
Genomic structure and the nature of a rearrangement of the 5q.13.2 telomeric part were determined in all analyzed subjects based on the copy number of 5q13.2 genes obtained by the MLPA analysis. In 66 out of 99 patients, reconstruction was aided by segregation analysis. Of the remaining 33 patients 13 were type II and 20 were type III cases. We were able to unmistakably reconstruct their 5q13.2 region as they carried only an increased SMN2 copy number (three or four copies) and no rearrangement of any surrounding gene, as suggested by their normal copy number determined by the MLPA analysis. Therefore, SMN1-to-SMN2 conversion was unambiguously the only rearrangement observed.
We observed 20 different alleles of the 5q13.2 telomeric part among 442 analyzed chromosomes (Figure 1). Thirteen alleles associated with the disease, found in patients and carriers, were grouped into six types (A–F) depending on the size of a rearrangement estimated through the involvement of the SERF1A and NAIP genes. In types A–D, the SMN1 gene was completely absent, while in types E and F the SMN1 absence was partial. Each allele type was further subdivided based on whether the absence of the SMN1 gene was due to deletion (subtype 1) or conversion to SMN2 (subtype 2). Two alleles most commonly associated with the disease were A1 (with a large-scale deletion encompassing SERF1A, SMN1 and NAIP, 78 chromosomes) and D2a (harboring only a conversion of SMN1 to SMN2, 124 chromosomes). Alleles G–M were restricted to the parent group and carried an increased copy number of either SMN1 or NAIP (Figure 1).
Individual correlation of SMN2, SERF1A and NAIP copy number with childhood-onset SMA types
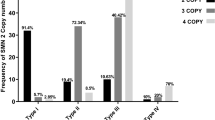
Strong inverse correlation was observed between the SMN2 copy number and childhood-onset types of SMA (Spearman rank test, ρ=0.85, P=2.2e−16) (Table 1 and Supplementary Figure 1). The majority of type I patients, 86.96% (20/23), carried two SMN2 copies, 4.35% (1/23) carried a single copy, while 8.69% (2/23) were found to have three copies. All type II cases carried three SMN2 copies. Among type III patients, 61.54% (24/39) were found to have four SMN2 copies, while 38.46% (15/39) carried three copies.
SERF1A copy number also showed strong inverse correlation with types of childhood-onset SMA (Spearman rank test, ρ=0.626, P=4.264e−10) (Table 1 and Supplementary Figure 1). In all, 9 out of 23 (39.13%) type I cases had no SERF1A copies, while such genotype was not detected in type II and type III patients. It is also noteworthy that none of type I patients carried all four SERF1A copies.
Inverse correlation between NAIP copy number and early-onset SMA type was also observed (Spearman rank test, ρ=0.523, P=2.722e−8) (Table 1 and Supplementary Figure 1). NAIP was homozygously deleted in 43.48% (10/23) type I patients, 8.11% (3/37) type II patients and only 2.56% (1/39) type III patients. Additionally, only 4.35% (1/23) of type I patients carried two NAIP copies, while frequencies of such genotype among type II and type III patients were 21.62% (8/37) and 53.85% (21/39), respectively.
Joint effect of the SMN2 and SERF1A genes on childhood-onset SMA types
Considering strong inverse correlations of individual CNP of SMN2, SERF1A and NAIP genes with early-onset SMA type, our next aim was to obtain a model explaining the phenotypic variation with the smallest set of predictor variables, which would at the same time test a joint effect of these genes. Patients were divided according to the pattern of homozygous deletion of SERF1A and/or NAIP genes into four groups (Table 2). SERF1A and/or NAIP genes were homozygously deleted only in type I patients, and these chromosome pairs carried two SMN2 copies, and a single copy in only one case (the first and the second rows in Table 2). The presence of at least one SERF1A and one NAIP copy was found among patients of all three SMA types, but in contrast to the previous patterns, this was detected in almost all type II and III cases and much less frequently in type I patients (the fourth row in Table 2). This pattern was mostly associated with an increased SMN2 copy number. The best generalized model explaining the variation in early-onset SMA phenotype with a minimal set of variables identified the main effect of SMN2 (P<2e−16) and SERF1A (P<2e−16) copy number and interaction of these two genes (P=0.02628) (Figure 2).

Joint effect of SMN2 and SERF1A on childhood-onset types of SMA. Patients with no SERF1A copies carrying either one or two SMN2 copies were diagnosed as type I. Remaining patients had at least one SERF1A copy, while the total number of SMN2 copies gradually increased up to four copies observed in type III patients. Homozygous absence of SERF1A is marked as ‘−’, while the presence of at least one SERF1A copy is marked as ‘+’. Dot size corresponds to SMN2 copy number.
Discussion
In this study, we determined the structure of 5q13.2 alleles in a group of 99 childhood-onset SMA patients of Serbian origin, carrying homozygous absence of the SMN1 gene, as well as in a group of 122 patients' parents. We showed that SMN2, SERF1A and NAIP copy number may individually modify the clinical outcome of early-onset SMA, but the best minimal model explaining the phenotypic variation of childhood-onset SMA revealed only SMN2 and SERF1A as major modifiers. Analyses of patients' parents showed the expected rate of de novo mutations and the expected frequency of SMN1 2/0 genotype among Serbian SMA carriers.
The inherent instability of the 5q13.2 segmental duplication was witnessed by the existence of 20 different telomeric alleles among 442 analyzed chromosomes. Homozygous absence of the entire SMN1 gene was confirmed in ~95% of analyzed patients. In the remaining cases one copy of SMN1 exon 8 was present, which is in concordance with general observation.15 The most frequently seen genomic rearrangements were large-scale deletions, encompassing the SMN1, SERF1A and/or NAIP genes, and gene conversion of SMN1 to SMN2, both of variable sizes. Large-scale deletions were mostly found in type I patients and rarely in type III, while SMN1-to-SMN2 gene conversion was mainly a characteristic of type II and III patients (Figure 1). Therefore, there is a significant difference in the meaning of homozygous absence of the SMN1 gene, which in type I patients almost exclusively means deleted, whereas in types II and III at least one SMN1 copy is converted to SMN2. The increase in NAIP and/or SMN1 copy number was seen only in alleles not associated with the disease.
The obtained correlation between SMN2 CNP and presenting childhood-onset SMA supports the previous evidence.12, 14, 15 In our study the majority of type I patients (~90%) carried two SMN2 copies, all type II patients carried three copies and type III patients carried three or four copies (~38% and ~62%, respectively). These results point to the conclusion that there is a relationship between SMN2 copy number and the nature of a 5q13.2 rearrangement. Two type I patients never gained the ability to sit unsupported despite having three SMN2 copies and three SERF1 copies, which indicates the involvement of other disease modifiers. Given that individuals with five or six copies of SMN2 develop very mild symptoms and are classified as type IV,15 and that eight SMN2 copies fully protect from developing SMA,28 it is not surprising that we did not detect any individual carrying more than four SMN2 copies.
Although the assessment of SMN2 copy number may have an important role in predicting disease severity and progression rate,14 not all SMN2 copies are functionally equivalent.12 Chimeric SMN2 copies with retained 5′ end of SMN1 are more capable of modifying the clinical outcome in comparison with copies in which a conversion encompasses the 5′ end of the SMN1 gene. We detected one chimeric copy in which only exon 7 was converted to SMN2, and one in which only exon 8 was SMN1, but the rest of the gene was SMN2 (Figure 1, alleles E2 and F2, respectively). However, both of them were found in type II patients. Intragenic SMN2 mutations, partial SMN2 deletions or duplications,14 and different degree of SMN2 promoter methylation29 may further modify the functionality of the SMN2 gene.
Observation that large-scale deletions are exclusive for type I patients20 is in line with our results showing homozygous deletion of telomeric SERF1A deletion only in this group of patients. Previous studies also demonstrated that SERF1A is more frequently deleted than NAIP and as frequently as SMN1 in type I SMA patients,22, 24 most probably because it lies closer to SMN1 than any other gene in the 5q13.2 region. It is interesting that deletion of the SERF1A gene was not found in any of 149 Chinese SMA patients with SMN1 homozygous deletion, including a group of 48 type I patients.25
According to the previous studies, the NAIP gene is absent in more than 50% of type I patients, but much less frequently in type II and type III cases.19, 21, 24, 25 This turned out to be relevant for our study in which homozygous absence of NAIP was almost exclusively seen in type I patients.
The best model describing the variability in childhood-onset SMA phenotype revealed the joint effect of two (SMN2 and SERF1A) out of three above-mentioned 5q13.2 genes showing individually significant correlation. Major effect of the increased SMN2 copy number is an expected scenario since it produces 10% of the functional SMN protein.13 The SMN2 gene is a target for promising antisense oligonucleotide-based therapy, directed to increasing SMN2 exon 7 inclusion and restoring the functional SMN protein.30 This therapy approach mediates partial phenotypic rescue in both mild and severe SMA mice models.30, 31 It is still unclear how the functional loss of SERF1A may enhance the SMA phenotype. SERF1A is a general regulator of protein aggregation, but it is unknown whether the protein misfolding has a role in SMA pathogenesis.17 It is also not known whether the functional loss of SERF1A gene may be partially compensated by its 5q13.2 centromeric paralog SERF1B.
An observed rate of SMN1 gene de novo mutations (~2%) in our study is similar to previous data32, 33 and may explain a high carrier frequency in the general population. However, germline mosaicism in the carriers without an identifiable mutation should be considered.34 In situations where a mutation is not seen in parents’ biological samples, paternal/maternal DNA test is useful for confirming the carrier status.
About 2–9% of carriers have two SMN1 copies on the same chromosome and none on the other (2/0 genotype).35 Therefore, any kind of SMN1 dosage analysis cannot differentiate between these carriers and individuals carrying two SMN1 copies, each on one chromosome (1/1 genotype). Only with segregation analysis we were able to distinguish three (2.46%) 2/0 carriers from carriers with a de novo mutation. If this scenario is encountered, then parents of a person suspected to be a 2/0 carrier should be included in the analysis, since usually one of them will possess one SMN1 copy (1/0 genotype), while the other will show three SMN1 copies at least (2/1 genotype).35
Conclusion
SMN2 and SERF1A show a joint effect in modifying childhood-onset SMA phenotype. Results of this study put the focus on the nature of 5q13.2 genomic rearrangements as a major determinant of their modyfing effects. Type I patients most frequently carry SMN1 homozygous deletion, no SERF1A copies and either one or two SMN2 copies. Type II and type III patients mainly posses SMN1 converted to SMN2 with at least one SERF1A copy, and the total number of SMN2 copies gradually increasing toward milder types. These data emphasize the significance of MLPA dosage analyses of the SMN2 and SERF1A genes, but it cannot be used to accurately determine early-onset SMA severity due to the existence of other disease modifiers. Dosage analyses of the SMN1 gene in both proband and their parents is necessary to detect 2/0 carriers and differentiate them from carriers of de novo mutation.
References
Pearn, J. Incidence, prevalence and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet. 15, 409–413 (1978).
Cusin, V., Clermont, O., Gerard, B., Chantereau, D. & Elion, J. Prevalence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counselling. J. Med. Genet. 40, e39 (2003).
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L. et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165 (1995).
Wirth, B., Herz, M., Wetter, A., Moskau, S., Hahnen, E., Rudnik-Schöneborn, S. et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am. J. Hum. Genet. 64, 1340–1356 (1999).
Munsat, T. L. & Davies, K. E. International SMA consortium meeting. (26-28 June 1992, Bonn, Germany). Neuromuscul. Disord. 2, 423–428 (1992).
Wang, C. H., Finkel, R. S., Bertini, E. S., Schroth, M., Simonds, A., Wong, B. et al. Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. J. Child. Neurol. 22, 1027–1049 (2007).
Wirth, B., Garbes, L. & Riessland, M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr. Opin. Genet. Dev. 23, 330–338 (2013).
Rochette, C. F., Gilbert, N. & Simard, L. R. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 108, 255–266 (2001).
Schmutz, J., Martin, J., Terry, A., Couronne, O., Grimwood, J., Lowry, S. et al. The DNA sequence and comparative analysis of human chromosome 5. Nature 431, 268–274 (2004).
Bailey, J. A. & Eichler, E. E. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat. Rev. Genet. 7, 552–564 (2006).
Girirajan, S., Campbell, C. D. & Eichler, E. E. Human copy number variation and complex genetic disease. Annu. Rev. Genet. 45, 203–226 (2011).
Burghes, A. H. When is a deletion not a deletion? When it is converted. Am. J. Hum. Genet. 61, 9–15 (1997).
Monani, U. R., Lorson, C. L., Parsons, D. W., Prior, T. W., Androphy, E. J., Burghes, A. H. et al. A single nucleotide difference that alters splicing patterns distinguishing the SMA gene and SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183 (1999).
Feldkötter, M., Schwarzer, V., Wirth, R., Wienker, T. F. & Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 70, 358–368 (2002).
Wirth, B., Brichta, L., Schrank, B., Lochmüller, H., Blick, S., Baasner, A. et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum. Genet 119, 422–428 (2006).
Lamar, K. M. & McNally, E. M. Genetic Modifiers for Neuromuscular Diseases. J. Neuromuscul. Dis. 1, 3–13 (2014).
van Ham, T. J., Holmberg, M. A., van der Goot, A. T., Teuling, E., Garcia-Arencibia, M., Kim, H. E. et al. Identification of MOAG-4/SERF as a regulator of age-related proteotoxicity. Cell 142, 601–612 (2010).
Liston, P., Roy, N., Tamai, K., Lefebvre, C., Baird, S., Cherton-Horvat, G. et al. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 379, 349–353 (1996).
Roy, N., Mahadevan, M. S., McLean, M., Shutler, G., Yaraghi, Z., Farahani, R. et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 80, 167–178 (1995).
Burlet, P., Bürglen, L., Clermont, O., Lefebvre, S., Viollet, L., Munnich, A. et al. Large scale deletions of the 5q13 region are specific to Werdnig-Hoffmann disease. J. Med. Genet. 33, 281–283 (1996).
Velasco, E., Valero, C., Valero, A., Moreno, F. & Hernández-Chico, C. Molecular analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy (SMA) families and correlation between number of copies of CBCD541 and SMA phenotype. Hum. Mol. Genet. 5, 257–263 (1996).
Scharf, J. M., Endrizzi, M. G., Wetter, A., Huang, S., Thompson, T. G., Zerres, K. et al. Identification of a candidate modifying gene for spinal muscular atrophy by comparative genomics. Nat. Genet. 20, 83–86 (1998).
Arkblad, E., Tulinius, M., Kroksmark, A. K., Henricsson, M. & Darin, N. A population-based study of genotypic and phenotypic variability in children with spinal muscular atrophy. Acta. Paediatr. 98, 865–872 (2009).
Amara, A., Adala, L., Ben Charfeddine, I., Mamaï, O., Mili, A., Lazreg, T. B. et al. Correlation of SMN2, NAIP, p44, H4F5 and Occludin genes copy number with spinal muscular atrophy phenotype in Tunisian patients. Eur. J. Paediatr. Neurol. 16, 167–174 (2012).
He, J., Zhang, Q. J., Lin, Q. F., Chen, Y. F., Lin, X. Z., Lin, M. T. et al. Molecular analysis of SMN1, SMN2, NAIP, GTF2H2, and H4F5 genes in 157 Chinese patients with spinal muscular atrophy. Gene. 518, 325–329 (2013).
Munsat, T. L. International SMA collaboration. Neuromuscul. Disord. 1, 81 (1991).
R Core Team R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2014).
Brahe, C. Copies of the survival motor neuron gene in spinal muscular atrophy: The more, the better. Neuromuscul. Disord. 10, 274–275 (2000).
Hauke, J., Riessland, M., Lunke, S., Eyüpoglu, I. Y., Blümcke, I., El-Osta, A. et al. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum. Mol. Genet. 18, 304–317 (2009).
Hua, Y., Sahashi, K., Hung, G., Rigo, F., Passini, M. A., Bennett, C. F. et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes. Dev. 24, 1634–1644 (2010).
Hua, Y., Sahashi, K., Rigo, F., Hung, G., Horev, G., Bennett, C. F. et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126 (2011).
Wirth, B., Schmidt, T., Hahnen, E., Rudnik-Schöneborn, S., Krawczak, M., Müller-Myhsok, B. et al. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am. J. Hum. Genet. 61, 1102–1111 (1997).
Ogino, S. & Wilson, R. B. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum. Genet. 111, 477–500 (2002).
Campbell, L., Daniels, R. J., Dubowitz, V. & Davies, K. E. Maternal mosaicism for a second mutational event in a type I spinal muscular atrophy family. Am. J. Hum. Genet. 63, 37–44 (1998).
Ben-Shachar, S., Orr-Urtreger, A., Bardugo, E., Shomrat, R. & Yaron, Y. Large-scale population screening for spinal muscular atrophy: clinical implications. Genet. Med. 13, 110–114 (2011).
Acknowledgements
This study was supported by a grant no. 173016 from the Ministry of Education, Science and Technological Development, Republic of Serbia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Brkušanin, M., Kosać, A., Jovanović, V. et al. Joint effect of the SMN2 and SERF1A genes on childhood-onset types of spinal muscular atrophy in Serbian patients. J Hum Genet 60, 723–728 (2015). https://doi.org/10.1038/jhg.2015.104
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.104

{kind=link}