
Abstract
Homozygous frameshift BRAT1 mutations were found in patients with lethal neonatal rigidity and multifocal seizure syndrome (MIM# 614498). Here, we report on two siblings with compound heterozygous mutations in BRAT1. They had intractable seizures from neonatal period, dysmorphic features and hypertonia. Progressive microcephaly was also observed. Initial electroencephalogram showed a suppression-burst pattern, leading to a diagnosis of Ohtahara syndrome. They both died from pneumonia at 1 year and 3 months, respectively. Whole-exome sequencing of one patient revealed a compound heterozygous BRAT1 mutations (c.176T>C (p.Leu59Pro) and c.962_963del (p.Leu321Profs*81)). We are unable to obtain DNA from another patient. The p.Leu59Pro mutation occurred at an evolutionarily conserved amino acid in a CIDE-N (N-terminal of an cell death-inducing DFF45-like effector) domain, which has a regulatory role in the DNA fragmentation pathway of apoptosis. Our results further support that mutations of BRAT1 could lead to epileptic encephalopathy.
Similar content being viewed by others

Main
BRAT1 at 7p22.3 encodes BRCA1-associated ATM (ataxia telangiectasia mutated) activator 1, which binds to both the tumor suppressing BRCA1 protein and ATM protein, and has important roles in sensing of DNA damaged lesions.1,2 Recently, homozygous frameshift mutations in BRAT1 have been reported to cause lethal neonatal rigidity and multifocal seizure syndrome (MIM# 614498) in three patients from three separate Amish sibships and another from a consanguineous Mexican family.3,4 Here, we present Japanese siblings with compound heterozygous mutations in BRAT1.
Case report
Case II-1
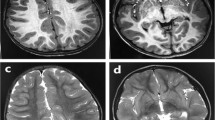
She was born to non-consanguineous Japanese parents as a first child without asphyxia after a 40-weeks' pregnancy (Figure 1a). Birth weight was 2644 g (−1.15 standard deviation (s.d.)) and head circumference (HC) 32.7 cm (−0.55 s.d.). Muscle hypertonia, micrognathia, short and webbed neck and dysmorphic face were observed at birth. Laboratory examination did not reveal any abnormalities. Serial seizures of her arms and legs twitching started at age 7 days, followed by generalized tonic clonic convulsions. Electroencephalography showed suppression-burst pattern, leading to diagnosis of Ohtahara syndrome or early myoclonic encephalopathy. Phenobarbital and clonazepam were not effective. Apnea attacks appeared at the age of 2 months. Zonisamide controlled her apnea attacks and general tonic convulsion at 8 months, but myoclonic seizures of the limbs and face were persistent. Her HC at 10 months is 43.0 cm (−2.3 s.d.). Serial brain magnetic resonance imaging revealed progressive cerebral and cerebellar atrophy (Figures 1b, c, e and f), and funduscopic examination revealed optic atrophy. She showed hypertonia, hyperreflexia and foot clonus, but no developmental milestones. She died of pneumonia at the age of 1 year and 9 months.

Clinical features of the family with Ohtahara syndrome. (a) A familial pedigree and BRAT1 mutations. T1-weighted (b, c) and T2-weighted (d) axial images through the basal ganglia, and T1-weighted (e–g) sagittal images. Progressive cerebral and cerebellar atrophy were observed in patient II-1, and mild atrophy and delayed white-matter myelination were observed in patient II-2. (h) Electroencephalography (EEG) at the age of 1 day showed the typical suppression-burst pattern in patient II-2. Histpathological features of a control female (i) and patient II-2 (j, k). A control female was born after 30 weeks’ gestation without asphyxia, and suffered from sudden death at home at the age of 3 months. Autopsy revealed only periventricular leukomalacia. (j) The cerebral cortex of frontal lobes showed remarkable loss of neurons in cortical layers IV and V compared with control case (i). The cortical neurons revealed neither dysgenesis nor abnormal substance accumulation inside. Kluver-Barrera's stain. (k) The cerebral white matter of frontal lobe showed gliosis (indicated by bidirectional arrow) and no myelination. Immunohistochemical stain of glial fibrillary acidic protein. Each bar indicates 500 μm.
Case II-2
She was born after 39 weeks’ gestation without asphyxia as a younger sister to case II-1. Her birth weight was 2540 g (−1.40 s.d.), height 45.5 cm (−1.84 s.d.) and HC 32 cm (−0.93 s.d.). Muscle hypertonia, generalized myoclonic seizures and partial clonic convulsions, poor voluntary movements, and dysmorphic features including bilateral talipes equinovarus, a round face, a thin lip and large ears were observed soon after birth. Exaggerated deep-tendon reflexes and Babinski reflex were also observed. Irritabilities and myoclonus of the limbs were readily induced by stimulation. Chromosomal analysis and laboratory examination showed no abnormalities.
In her clinical course, her various convulsions consisted of myoclonic, clonic and tonic seizures, which were worsen by phenytoin. Apnea attacks increased at day 53, which were relieved by zonisamide. Brain magnetic resonance imaging at 3 months showed mild cerebral and cerebellar atrophy, and delayed myelination of the cerebral white matter (Figures 1d and g). Initial electroencephalography at second day of birth showed suppression-burst pattern (Figure 1h). Optic atrophy was detected by funduscopy. She obtained no developmental milestones even without swallowing her own saliva, thereafter died of pneumonia at the age of 3 months. The weight and the HC were 4884 g (−1.6 s.d.) and 35 cm (−3.8 s.d.), respectively. An autopsy was performed.
Neuropathological findings of case II-2
The brain weight was 321 g, much smaller than that of age-matched control brain (718 g). The cerebellum was proportionately small. No cortical dysgenesis was seen, but cortical neurons were moderately depleted. Remaining neurons showed no substance accumulation inside (Figures 1i and j). The entire white matter of the cerebrum showed moderate gliosis and no myelination in the frontal lobe (Figure 1k). The moderate Purkinje cells depletion and some dendritic expansions were observed in the cortical layer of the cerebellum. The neurons of the dentate nucleus were also depleted. No calcification was found in the caudate and globus pallidus but tiny microglial nodule in the thalamus. No specific neuronal loss was observed in hippocampal area. Neuronal cells of the brain stem, including pontine nuclei and olivary nuclei, were preserved relatively. Myelination of brain stem and spinal cord was developed well.
Results and Discussion
Genomic DNA of liver tissues of case II-2 was captured using the SureSelect Human All Exon v5 Kit (Agilent Technologies, Santa Clara, CA, USA), and sequenced with on HiSeq2000 (Illumina, San Diego, CA, USA) with 101 bp paired-end reads. Exome data processing was performed as previously described.5 Coverage data and variant filtering are shown in Supplementary Table S1. Among 19 genes possessing two heterozygous variants (possible compound heterozygous mutations) or a homozygous mutation based on the autosomal recessive model, we found two mutations in BRAT1. Compound heterozygosity of the two mutations was confirmed by Sanger sequencing: c.176T>C (p.Leu59Pro) and c.962_963del (p.Leu321Profs*81) were transmitted from her father and mother, respectively. These two mutations were not found in the ESP6500 exomes or among our 575 in-house control exomes. The p.Leu59Pro mutation was predicted as damaging by sorting intolerant from tolerant (SIFT), PolyPhen2 and MutationTaster (Supplementary Table S2). All experimental protocols used were approved by the Institutional Review Board of Yokohama City University School of Medicine.
BRAT1, also donated as BAAT1, is identified as a binding protein with BRCA1 and ATM. The role of BRAT1 is under investigation, but is considered to have roles in sensing of DNA damaged lesions and apoptosis.1,2 To date, two homozygous frameshift mutations (c.638dupA and c.453_454insATCTTCTC) have been reported in a total of four families in which three Amish families were likely to be related (Figure 2).3,4 The affected individuals including our cases commonly showed hypertonia, intractable seizures, microcephaly and early lethality. Although we are unable to check the BRAT1 mutations in case II-1 as biological samples were unavailable, these striking similarities of clinical features suggest that she also had two BRAT1 mutations, which are highly likely to be pathogenic. It is interesting to note that the p.Leu59Pro mutation occurred in an evolutionarily conserved (chemically similar) amino acid within N-terminal of an cell death-inducing DFF45-like effector (CIDE-N) domain (Figure 2). Indeed, the CIDE-N domain is known to have a regulatory role in the DNA fragmentation pathway of apoptosis.6 Considering the fact that progressive atrophy and neuronal cell loss were observed in cerebrum and cerebellum, aberration of apoptosis may be one of pathologies caused by BRAT1 mutations.

Schematic representation of BRAT1 mutations. BRAT1 protein consists of 821 amino-acid residues with one CIDE-N (N-terminal of an cell death-inducing DFF45-like effector) domain and two HEAT (Huntingtin, Elongation factor 3, A subunit of protein phosphatase 2A, and TOR1) repeat domains.2 Leu59 is located within the CIDE-N domain, and is relatively conserved among vertebrates as only chemically similar amino acids are seen at this position. Multiple amino-acid sequences of BRAT1/Brat1 proteins were aligned using the CLUSTALW website. Two previously reported mutations are shown above BRAT1 and two mutations found in this study are below the protein.
In conclusion, we describe two Japanese girls with Ohtahara syndrome, possessing compound heterozygous BRAT1 mutations. Our report suggests that regulation of apoptosis by BRAT1 may be important for normal brain development.
References
Aglipay J. A., Martin S. A., Tawara H., Lee S. W., Ouchi T. ATM activation by ionizing radiation requires BRCA1-associated BAAT1. J. Biol. Chem. 281, 9710–9718 (2006).
Ouchi M., Ouchi T. Regulation of ATM/DNA-PKcs phosphorylation by BRCA1-Associated BAAT1. Genes Cancer 1, 1211–1214 (2010).
Saunders C. J., Miller N. A., Soden S. E., Dinwiddie D. L., Noll A., Alnadi N. A. et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 4, 154ra135 (2012).
Puffenberger E. G., Jinks R. N., Sougnez C., Cibulskis K., Willert R. A., Achilly N. P. et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE 7, e28936 (2012).
Saitsu H., Nishimura T., Muramatsu K., Kodera H., Kumada S., Sugai K. et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449 (2013).
Lugovskoy A. A., Zhou P., Chou J. J., McCarty J. S., Li P., Wagner G. . Solution structure of the CIDE-N domain of CIDE-B and a model for CIDE-N/CIDE-N interactions in the DNA fragmentation pathway of apoptosis. Cell 99, 747–755 (1999).
Acknowledgements
We would like to thank the patient and her family for their participation in this study. We thank Nobuko Watanabe for her technical assistance. This study was supported by: the Japanese Ministry of Health, Labour, and Welfare; the Japan Society for the Promotion of Science (a Grant-in-Aid for Scientific Research (B) (25293085, 25293235), a Grant-in-Aid for challenging Exploratory Research (26670505); a Grant-in-Aid for Scientific Research (A) (13313587)); the Takeda Science Foundation; the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems; the Strategic Research Program for Brain Sciences (11105137); and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (12024421).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Saitsu, H., Yamashita, S., Tanaka, Y. et al. Compound heterozygous BRAT1 mutations cause familial Ohtahara syndrome with hypertonia and microcephaly. J Hum Genet 59, 687–690 (2014). https://doi.org/10.1038/jhg.2014.91
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.91
This article is cited by
-
BRAT1–related disorders: phenotypic spectrum and phenotype-genotype correlations from 97 patients
European Journal of Human Genetics (2023)
-
Novel variant in BRAT1 with the lethal neonatal rigidity and multifocal seizure syndrome
Pediatric Research (2022)
-
An intronic variant in BRAT1 creates a cryptic splice site, causing epileptic encephalopathy without prominent rigidity
Acta Neurologica Belgica (2020)
-
Targeted next-generation sequencing analysis in couples at increased risk for autosomal recessive disorders
Orphanet Journal of Rare Diseases (2018)
-
The Impact of Next-Generation Sequencing on the Diagnosis and Treatment of Epilepsy in Paediatric Patients
Molecular Diagnosis & Therapy (2017)
