
Abstract
Kinesins are a large superfamily of molecular motors. They move along microtubule filaments and are powered by the hydrolysis of ATP. This transport system is essential for neuronal function and survival. KIF1A belongs to the kinesin 3 family and involves in the anterograde transport of synaptic vesicle precursors along axons. Several studies confirmed that KIF1A mutations cause spastic paraplegia and sensory neuropathy in an autosomal-recessive fashion. A missense mutation in the KIF1A gene (p.Thr99Met) has been reported in a patient with intellectual disability (ID), axial hypotonia and peripheral spasticity. Mild atrophy of the cerebellar vermis was found on magnetic resonance imaging. The mutation was heterozygous and de novo. We identified the second patient with the p.T99M mutation in the KIF1A gene by whole-exome sequencing. He showed severe ID, spasticity, optic atrophy, neurogenic bladder, growth failure and progressive cerebellar atrophy. The p.T99M mutation may be a common recurrent mutation. We suppose that this specific mutation of KIF1A shows a novel neurodegenerative syndrome.
Similar content being viewed by others


Introduction
Kinesins are a large superfamily of molecular motors. Kinesins move along microtubule filaments and are powered by the hydrolysis of ATP. KIF1A (MIM*601255) encodes a motor protein gene involved in the anterograde transport of synaptic vesicle precursors along axons.1 Knockout of Kif1a in mice is lethal soon after birth due to massive axonal and neuronal cell body degeneration in the central nervous system.2 KIF1A transports a synaptic vesicle precursor and KIF1A-mediated axonal transport has a critical role in the viability, maintenance and function of neurons, particularly mature neurons.2
Abnormalities in the axonal transport of synaptic vesicles due to KIF1A mutations cause spastic paraplegia and sensory neuropathy in an autosomal-recessive fashion.3, 4, 5 Hamdan et al.6 identified a missense mutation (p.Thr99Met) in the KIF1A gene with whole-exome sequencing (WES) in a patient with non-syndrome intellectual disability (ID). The mutation was heterozygous and de novo. We identified the second patient with the p.Thr99Met mutation.
Materials and methods
With the approval of our institutional ethics committee, we planned a proband-parent trio approach using WES (Supplementary Methods). We generated 6.54 Gb sequence on average. The average read depth of the on-target regions was 53.4. To check the quality of our WES base calls, we genotyped eight samples using both next-generation sequencing (NGS) and single nucleotide polymorphism (SNP) array and compared them. On average, the genotypes for 244 698 locations were comparable between the two data sets and 99.95% genotypes were concordant between our NGS calls and the genotype of the SNP array (Supplementary Table S1). We validated the 140 mismatched positions for the NA18943 sample by SureSelect V4 (Agilent Technologies, Santa Clara, CA, USA) that were discordant using a Sanger sequencing (Supplementary Tables S2 and S3). As a result, 77 of 140 genotypes were consistent with our NGS calls. On the basis of this validation, we estimated the false positive and false negative rates of our calling methods to be 0.021% (50/243 573) and 0.064% (13/20 424), respectively. Note that these rates are conservative estimations, as the single nucleotide variants (SNVs) that could not be successfully determined using Sanger sequencing were counted as mismatched. In addition, the test sample, NA18943, had the most discordant SNP calls of the set.
Results
Clinical report
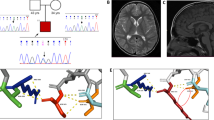
The 8-year-old boy was the third child of healthy and non-consanguineous Japanese parents. After an uncomplicated pregnancy, he was born at 40 weeks of gestation. His birth weight was 3700 g, length was 51 cm and head circumference was 34 cm. He was hypotonic and his developmental milestones were markedly delayed. Routine laboratory tests revealed mildly elevated transaminases. Aspartate aminotransferase ranged from 77 to 170 IU l−1 (normal <30 IU l−1), whereas alanine transaminase ranged from 58 to 206 IU l−1 (normal <30 IU l−1) during infantile period. G-banded chromosome analysis was normal. Brain magnetic resonance imaging at 8 months of age revealed mildly enlarged lateral ventricles, hypoplasia of the corpus callosum, thin pituitary gland and atrophic cerebellar vermis (Figures 1 and 2).

Axial T1-weighted magnetic resonance image of patient ((a) 1 year of age and (b) 4 years of age) showed enlarged lateral ventricles and decreased white matter volume. Lateral ventricles slightly became larger.

Sagittal T1-weighted magnetic resonance image of patient ((a) 1 year of age, (b) 3 years of age and (c) 4 years of age) showed hypoplasia of corpus callosum and cerebellar atrophy. Cerebellar vermis was atrophic (arrow).
Physical examination identified no dysmorphic features. He showed axial hypotonia and peripheral spasticity. Deep tendon reflexes were exaggerated. Nystagmus was noted. Ocular pursuit was poor. Ophthalmological investigation revealed optic atrophy. Visual evoked potentials showed a poor response. Head control was achieved at 18 months of age. He rolled over at 2 years of age and could sit without support at 3 years of age. His height gain was under −3 s.d., and complete isolated growth hormone deficiency was revealed. Replacement therapy was started from 3 years of age. Muscle biopsy gave nonspecific findings. Electromyograms with nerve conduction velocity studies revealed normal findings.
Generalized tonic-clonic convulsion appeared at 4 years of age. Electroencephalography (EEG) showed diffuse small spikes. His locomotive activity gradually decreased. Axial hypotonia and peripheral spasticity became more apparent. His lower extremities were spastic and his left hip joint was dislocated. His height was 106 cm (−1.6 s.d.) and weight was 14.7 kg (−1.9 s.d.). His head circumference was 47.5 cm (−2.4 s.d.) at 6 years of age. He had no cognitive and language perception. Obstructive sleep apnea appeared. He developed repeated episodes of urinary retention. Ultrasonography of the urinary tract showed a distended bladder with normal kidneys and he was diagnosed with neurogenic bladder. He was started on clean intermittent catheterization to ensure adequate bladder drainage. He also showed chronic constipation. Repeated neuroradiological investigations showed progressive cerebral and cerebellar atrophy (Figures 1 and 2).
Whole-exome sequencing
A de novo missense mutation, NM_001244008:exon4:c.C296T:p.T99M, was identified in KIF1A. This mutation was confirmed by Sanger sequencing.
Discussion
Hamdan et al.6 reported a patient with p.T99M mutation in the KIF1A gene. The patient was a 3-year-old girl with severe ID, axial hypotonia and peripheral spasticity. Mild atrophy of the vermian region of the cerebellum was found. The patient showed similar conditions to our patient. Our patient showed progressive cerebral and cerebellar atrophy. The progressive nature of KIF1A mutations can be explained by the evidence that mature neurons are particularly affected.2 Optic atrophy and neurogenic bladder were novel findings in our patient. We supposed that p.T99M mutation may affect optic nerve and autonomic nervous system. Complete growth hormone (GH) deficiency might be a coincidental complication.
Hamdan et al.6 studied the impact of p.T99M on KIF1A subcellular localization in neurites. They transfected primary rat hippocampal neurons with KIF1A motor domain–EGFP fusion constructs. Wild-type KIF1A accumulated in distal regions of neurites, whereas KIF1A molecule with the p.T99M mutation showed greatly reduced distal localization and increased accumulation throughout the cell body and proximal neurites.6 Because the T99 residue lies in the conserved ATP-binding p loop of the motor domain of KIF1A, the mutation may affect KIF1A to obtain energy from ATP. As KIF1A can homodimerize, heterozygous mutations may cause a dominant negative effect.
It is interesting to investigate patients with neurodegeration of unknown cause for p.T99M mutation, which may be a common recurrent mutation. We propose that this specific mutation of KIF1A shows a novel neurodegenerative syndrome.
References
Okada Y ., Yamazaki H ., Sekine-Aizawa Y ., Hirokawa N . The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonaltransport of synaptic vesicle precursors. Cell 1995; 81: 769–780.
Yonekawa Y ., Harada A ., Okada Y ., Funakoshi T ., Kanai Y ., Takei Y . et al. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J. Cell Biol. 1998; 141: 431–441.
Erlich Y ., Edvardson S ., Hodges E ., Zenvirt S ., Thekkat P ., Shaag A . et al. Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res. 2011; 21: 658–664.
Rivière J. B ., Ramalingam S ., Lavastre V ., Shekarabi M ., Holbert S ., Lafontaine J . et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am. J. Hum. Genet. 2011; 89: 219–230.
Klebe S ., Lossos A ., Azzedine H ., Mundwiller E ., Sheffer R ., Gaussen M . et al. KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur. J. Hum. Genet. 2012; 20: 645–649.
Hamdan F. F ., Gauthier J ., Araki Y ., Lin D. T ., Yoshizawa Y ., Higashi K . et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 2011; 88: 306–316.
Acknowledgements
This study was supported by the Health and Labour Research Grants in 2013 by Ministry of Health, Labour and welfare in Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Okamoto, N., Miya, F., Tsunoda, T. et al. KIF1A mutation in a patient with progressive neurodegeneration. J Hum Genet 59, 639–641 (2014). https://doi.org/10.1038/jhg.2014.80
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.80
This article is cited by
-
Four pedigrees with aminoacyl-tRNA synthetase abnormalities
Neurological Sciences (2022)
-
Monoallelic KIF1A-related disorders: a multicenter cross sectional study and systematic literature review
Journal of Neurology (2022)
-
KIF1A-related autosomal dominant spastic paraplegias (SPG30) in Russian families
BMC Neurology (2020)
-
Presynaptic disorders: a clinical and pathophysiological approach focused on the synaptic vesicle
Journal of Inherited Metabolic Disease (2018)
-
Siblings with optic neuropathy and RTN4IP1 mutation
Journal of Human Genetics (2017)
