
Abstract
Cornelia de Lange syndrome (CdLS) is a clinically and genetically heterogeneous developmental disorder. The clinical features of CdLS include growth retardation, intellectual disability, limb defects, typical facial dysmorphism and other systemic involvement. Here, we present the clinical and genetic characterization of a sporadic CdLS trio. The proband is a 7-year-old girl with typical CdLS, and both parents are apparently healthy. Whole-exome sequencing of the patient and of both her unaffected parents revealed a previously unobserved de novo mutation in exon 6 of the HDAC8 gene (chrX: 71684483, c.586 A>T; p.M196K). Thus, we have further founded that the p.M196K mutation in HDAC8 is a relevant causal mutation for CdLS.
Similar content being viewed by others


Introduction
Cornelia de Lange syndrome (CdLS, MIM #610759, 300882, 300590, 614701 and 122470) is a congenital multisystemic genetic disorder with variable clinical expression.1 It is characterized by typical facial dysmorphism, growth retardation, intellectual disability, limb defects, ocular disorders and other systemic involvement.1, 2 The worldwide prevalence of CdLS is estimated to be between 1:10 000 and 1:30 000 live births.3
Most CdLS cases are due to spontaneous genetic mutations. To date, mutations in three core cohesin subunits (SMC1A, SMC3 and RAD21) and in two cohesin-regulatory proteins (NIPBL and HDAC8) have been identified in CdLS probands.3 NIPBL, which is located on chromosome 5p13 and encodes a cohesin regulatory protein, is the most common genetic cause of CdLS. Heterozygous mutations in NIPBL are found in at least 60% of CdLS probands.4, 5 Mutations in the core cohesion complex proteins, encoded by the SMC1A, SMC3 and RAD21 genes, together account for ∼5% of subjects, often with atypical CdLS features.6, 7, 8 Recently, mutations in HDAC8 have been found to be associated with CdLS. The HDAC8 gene encodes the lysine deacetylase for the cohesin subunit SMC3 in humans. Mutations in HDAC8 are found in ∼5% of individuals with CdLS.9, 10
Here, we report a patient with typical CdLS possessing a previously unreported de novo HDAC8 mutation that was identified via trio exome sequencing.
Materials and Methods
Case report
A 7-year-old girl was brought to our eye clinic with a confirmed diagnosis of CdLS for ocular anomalies. She was the firstborn child of healthy nonconsanguineous parents, and her family history was negative regarding genetic disease. She was delivered at 41 weeks of gestational age by cesarean section because of maternal problems. Her birth weight was 3.5 kg, birth length 52 cm and head circumference 35 cm at 33 days after birth. She had a documented history of developmental delay and exhibited characteristic facial features: synophrys, long curly eyelashes, a depressed nasal bridge, anteverted nares (Figure 1a) and widely spaced teeth. Her hands and feet were small, and she exhibited brachydactyly in her forefinger, midfinger and pinkie finger (Figure 1b). Upon ophthalmic examination, cycloplegic refraction demonstrated severe myopia (−10.25 D) in the right eye but no refractive error in the left eye. She was also diagnosed with myopia, anisometropia and amblyopia.

Features of the proband. (a) Characteristic facial features of the proband, including synophrys, long curly eyelashes, a depressed nasal bridge, anteverted nares and widely spaced teeth. (b) Upper-limb anomalies observed in the patient, including brachydactyly in her forefinger, midfinger and pinkie finger.
Whole-exome sequencing
Genomic DNA was isolated from peripheral blood leukocytes using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany), captured using the Truseq Human All Exome 62 Mb kit (Illumina, San Diego, CA, USA) and sequenced on an Illumina HiSeq2000 (Illumina), generating 2 × 100 bp paired-end reads. Exome data processing, variant calling and variant annotation were performed as previously described.11 From these variants, we identified putative de novo mutations as sites where both parents were homozygous for the reference sequence, while the offspring was heterozygous, without considering the genotype depth for each candidate variant.
Sanger sequencing
The putative de novo mutations were validated via Sanger sequencing of the carrier and both parents. The PCR primers for the 16 putative de novo mutations are summarized in Supplementary Table 1. Sequencing was performed using BigDye Terminator v3.1 cycle sequencing, and the obtained sequences were analyzed on an ABI 3730 DNA Analyzer (Applied Biosystems, Carlsbad, CA, USA).
Prediction of functional impacts
The pathogenicity of the identified missense mutations was tested using PROVEAN12 (http://provean.jcvi.org/index.php), SIFT13 (http://sift.jcvi.org/) and Mutation Taster14 (http://www.mutationtaster.org/) online software.
Analysis of evolutionary conservation
Data on HDAC8 orthologous genes were downloaded from the homologene database (http://www.ncbi.nlm.nih.gov/homologene). Multiple-sequence alignment was carried out using ClustalW (http://www.ebi.ac.uk/clustalw).15
Results
For each sample, >8.5 Gbp of raw data were obtained through exome sequencing, showing a mean coverage of >50-fold (see Supplementary Table 2), and a total of 143 177 SNVs were detected. The exome sequencing data covered all exons of known CdLS-related genes (SMC1A, SMC3, RAD21, NIPBL and HDAC8), and no known functional mutations were identified in these genes (see Supplementary Table 3). For de novo mutation filtering, 16 putative de novo mutations were identified according to the standard that both parents were homozygous for the reference sequence and the offspring was heterozygous (see Supplementary Table 4). Only two sites were confirmed to be totally consistent with the exome sequencing data of each family member (KPRP, rs17612167; HDAC8, c.586 A>T, p.M196K) (Figure 2a and Supplementary Figure 1). All the other 14 variants were unconfirmed because all these variants showed homozygosity in the exome sequencing data of one of the parents and heterozygosity in Sanger sequencing. The single-nucleotide polymorphism (SNP) rs17612167 (A/T) was further excluded because it is a common SNP in the Chinese population (minor allele frequency (MAF)=0.189). Moreover, by using Sanger Sequencing, the p.M196K mutation in HDAC8 was detected to be absent in 250 among the unrelated Chinese control population. Therefore, we further confirmed that the de novo mutation (c.586 A>T, p.M196K) in HDAC8 was the causal mutation in the proband of this CdLS trio.

Mutation in HDAC8. (a) Sequence of the de novo HDAC8 gene mutation (c.586 A>T, p.M196K) in all subjects in the sporadic CdLS trio. (b) Evolutionary conservation analysis of the de novo HDAC8 gene mutation (c.586 A>T, p.M196K).
In addition, the p.M196K mutation was predicted to be pathogenic by the algorithms of PROVEAN, SIFT and Mutation Taster (categorized as ‘deleterious,’ ‘damaging’ and ‘disease causing,’ respectively). The identified mutation (p.M196K) is highly conserved throughout evolution across species (Figure 2b).
Discussion
In this study, we identified two de novo mutations (KPRP, rs17612167; HDAC8, c.586 A>T, p.M196K) in a CdLS trio by both exome sequencing and Sanger sequencing. There are two reasons that rs17612167 was excluded from the candidate causal mutations of CdLS: first, rs17612167 is a common SNP with a MAF of 0.189 in the Chinese population; second, although the A>T base change of rs17612167 variant can result in a p. Q14H mutation in KPRP gene, the effect of this mutation was predicted to be moderate on the gene function. Hence, according to the results in the present study, the de novo p.M196K mutation in HDAC8 was the most recognized candidate variant associated with CdLS.
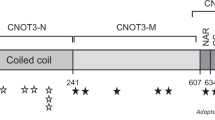
Recently, Deardorff et al.10 identified four HDAC8 mutations in six individuals with typical CdLS or an overlapping phenotype. Subsequently, Kaiser et al.9 screened an internationally assembled cohort of 586 patients with typical CdLS and overlapping clinical presentations and found 35 affected individuals whom they identified as having mutations in HDAC8. In both studies, the authors found six different chromosomal microdeletions or microduplications, three nonsense mutations, one splice site and 16 different missense mutations (p.I19S, p.H42P, p.H71Y, p.P91L, p.G117E, p.C153F, p.H180R, p.A188T, p.D233G, p.D237Y, p.K239-Y240N, p.I243N, p.G304R, p.T311M, p.G320R and p.H334R) (Figure 3). Thus, this is the first study to identify the de novo p.M196K mutation in HDAC8 in a sporadic CdLS patient (Figure 3). HDAC8, which is located on chromosome Xq13.1, encodes a histone deacetylase that deacetylates SMC3, which has been removed from chromatin to establish the cohesiveness of chromatin-loaded cohesin during both prophase and anaphase. Both Deardorff et al. and Kaiser et al. found that HDAC8 mutations in CdLS patients could cause reduced HDAC8 enzymatic activity. According to the structure of the HDAC protein (Figure 3), most of the CdLS-related mutations detected in the previous studies are located at the α helix and β strand’s second structure region; the p.M196K mutation is just located at a β-strand region of HDAC8. M196 is in a hydrophobic environment, and a mutation resulting in a larger, partially hydrophilic residue might subtly affect the packing interactions. Thus, the identified mutation (p.M196K) may also reduce HDAC8 deacetylase activity, which should be further functionally studied.

Schematics of protein alterations in HDAC8 detected by the previous and present study. A helix in HDAC8 is colored blue, the β strand in HDAC8 is colored red. A full color version of this figure is available at the Journal of Human Genetics journal online.
In summary, we applied whole-exome sequencing and functional prediction methods in this study and identified a de novo mutation (c.586 A>T, p.M196K) in HDAC8 in our sporadic CdLS trio that has not been detected in previous studies. Thus, we have further confirmed that HDAC8 is a relevant gene for CdLS.
References
Barakat, M. R., Traboulsi, E. I. & Sears, J. E. Coats' disease, megalopapilla and Cornelia de Lange syndrome. Ophthalmic Genet. 30, 106–108 (2009).
Milot, J. & Demay, F. Ocular anomalies in de Lange syndrome. Am. J. Ophthalmol. 74, 394–399 (1972).
Mannini, L., Cucco, F., Quarantotti, V., Krantz, I. D. & Musio, A. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Hum. Mutat. 34, 1589–1596 (2013).
Krantz, I. D., McCallum, J., DeScipio, C., Kaur, M., Gillis, L. A., Yaeger, D. et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 36, 631–635 (2004).
Tonkin, E. T., Wang, T. J., Lisgo, S., Bamshad, M. J. & Strachan, T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 36, 636–641 (2004).
Deardorff, M. A., Kaur, M., Yaeger, D., Rampuria, A., Korolev, S., Pie, J. et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 80, 485–494 (2007).
Musio, A., Selicorni, A., Focarelli, M. L., Gervasini, C., Milani, D., Russo, S. et al. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 38, 528–530 (2006).
Chen, J. M., Fichou, Y., Jamet, D., Dupont, I., Cooper, D. N., Le Maréchal, C. et al. Small deletions within the RHD coding sequence: a report of two novel mutational events and a survey of the underlying pathophysiologic mechanisms. Transfusion 53, 206–210 (2013).
Kaiser, F. J., Ansari, M., Braunholz, D., Concepción Gil-Rodríguez, M., Decroos, C., Wilde, J. J. et al. Loss of function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X linked inheritance. Hum. Mol. Genet. 23, 2888–2900 (2014).
Deardorff, M. A., Bando, M., Nakato, R., Watrin, E., Itoh, T., Minamino, M. et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489, 313–317 (2012).
O'Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7, e46688 (2012).
Ng, P. C. & Henikoff, S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Reva, B., Antipin, Y. & Sander, C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 8, R232 (2007).
Li, K. B. ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics 19, 1585–1586 (2003).
Acknowledgements
We thank the patient’s family for their participation in this study. This work was supported by the National Natural Science Foundation of China (31200954, Beijing, PR China), a China Postdoctoral Science Foundation-funded project (2012M510110 and 2013T60440, Beijing, PR China), and Foundation of Zhejiang Educational Committee (Y201330229, PR China).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Feng, L., Zhou, D., Zhang, Z. et al. Exome sequencing identifies a de novo mutation in HDAC8 associated with Cornelia de Lange syndrome. J Hum Genet 59, 536–539 (2014). https://doi.org/10.1038/jhg.2014.60
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.60
This article is cited by
-
Understanding the Landscape of X-linked Variants Causing Intellectual Disability in Females Through Extreme X Chromosome Inactivation Skewing
Molecular Neurobiology (2020)
-
Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement
Nature Reviews Genetics (2018)
-
A commentary on exome sequencing identifies a de novo mutation in HDAC8 associated with Cornelia de Lange syndrome
Journal of Human Genetics (2014)

{kind=link}