
Abstract
We report on a 1-year-old boy with microcephaly with a simplified gyral pattern, early-onset seizures, congenital hearing loss and a severe developmental delay. Trio-based whole-exome sequencing identified candidate compound heterozygous mutations in two genes: c.163G>T (p.Ala55Ser) and c.874G>A (p.Gly292Arg) in polynucleotide kinase 3′-phosphatase gene (PNKP), and c.195G>A (p.Met65Ile) and c.1210A>C (p.Ser404Arg) in PCDH15. PNKP and PCDH15 mutations have been reported in autosomal recessive microcephaly with early-onset seizures and developmental delay syndrome, and Usher syndrome type 1F, respectively. Our patient showed neurological features similar to reported cases of both syndromes that could be explained by the observed mutations in both PNKP and PCDH15, which therefore appear to be pathogenic in this case.
Similar content being viewed by others

Main
Central nervous systems suffer from various types of DNA damage, so DNA repair systems are indispensable for the correct proliferation and differentiation of the brain. DNA repair deficiency syndromes show different degrees of neurological impairment including microcephaly, neuropathy and neurodegeneration.1 Mutations in the polynucleotide kinase 3′-phosphatase gene (PNKP), encoding an enzyme involved in the DNA repair pathway, have been implicated in autosomal recessive microcephaly with early-onset intractable seizures and developmental delay (MCSZ, OMIM #613402) syndrome,2, 3 while protocadherin-15 gene (PCDH15) mutations cause Usher syndrome, type 1F. Here, we describe a Japanese patient with microcephaly with a simplified gyral pattern, early-onset seizures, severe developmental delay and congenital hearing loss, and identify pathogenic mutations in PNKP and PCDH15.
Case report
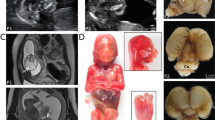
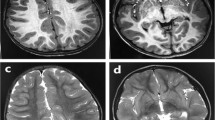
A Japanese boy was born to nonconsanguineous healthy parents as a second child after an uneventful 39-week pregnancy (Figure 1a). He presented with microcephaly (head circumference, 25.5 cm, −4.7 standard deviation (s.d.)) and growth restriction. His birth weight was 2114 g (−2.9 s.d.) and length was 44.5 cm (−2.5 s.d.). No other dysmorphic features or organ abnormalities were recognized. He failed the newborn hearing screening test twice. His seizures and hypertonus began at 1 month of age. He has exhibited frequent complex partial seizures with apnea since the age of 6 months and his electroencephalogram showed focal spikes. Brain magnetic resonance imaging at 16 days after birth showed microcephaly with a simplified gyral pattern and cerebellar hypoplasia (Figure 1b). Both the white matter volume and corpus callosum were reduced in size proportionally to the cerebrum. His feeding problems necessitated tubing after birth, and subsequently gastrostomy and Nissen fundoplication surgery were performed at the age of 9 months because of gastroesophageal reflux. At 17 months, his length was 67 cm (−4.2 s.d.), weight 6.5 kg (−3.3 s.d.) and head circumference 34 cm (−8.1 s.d.). He was able to hold his head, sometimes follow things with his eyes and smile, but could not roll over, sit independently or say any meaningful words. He presented with muscle hypotonia with limited lower leg movement, but muscle tonus varied depending on his temper. Nerve conduction studies were normal. His seizures were well controlled on phenobarbital and levetiracetam. Auditory steady-state response testing indicated mild to moderate hearing loss. External auditory meatus and tympanic membrane showed no anatomical changes by physical examination. The intracranial imaging examination was not performed because sedation may worsen his general condition. Therefore his hearing impairment remained elusive. G-banded analysis showed a normal karyotype (46,XY). Tests for metabolic disorders and TORCH were all negative. Experimental protocols for genetic analysis were approved by the Institutional Review Board of Yokohama City University School of Medicine. Clinical information and peripheral blood samples were acquired from family members after receiving their written informed consent.

(a) Familial pedigree and PNKP (red) and PCDH15 (blue) mutations. WT, wild-type allele. (b) Brain magnetic resonance imaging (MRI) of the patient at 16 days after birth (T2 weighted images). Microcephaly, a simplified gyral pattern with normal cortical thickness, slightly enlarged ventricles, cerebellar hypoplasia, and proportional reduction of white matter volume with a thin corpus callosum are evident. (c) Schematic presentation of PNKP protein and location of mutations. Five reported mutations are depicted above the PNKP protein. Two novel mutations (A55S and G292R) found in this study are shown below the protein. Three boxes indicate functional domains. A full color version of this figure is available at the Journal of Human Genetics journal online.
Whole-exome sequencing
DNA was extracted from the peripheral blood of the patient and his parents. DNA was captured with the SureSelect Human All Exon V5 Kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina Hiseq2000 (Illumina, San Diego, CA, USA) with 101-bp paired-end reads. Exome data processing, variant calling and variant annotation were performed as previously described.4 Prediction of significance in variants was performed by Polyphen-25 and MutationTaster software.6 Trio analysis identified candidate variants meeting a de novo occurrence model and autosomal and X-linked recessive models. All candidate variants were confirmed by Sanger sequencing using ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Results and Discussion
A total of 369 rare protein-altering and splice-site variants were detected in the patient. No candidate de novo variants were found when variants were excluded in unaffected parents, dbSNP137 data, 6500 Exome Sequencing Project (ESP) exome databases and 408 in-house exomes. Variants consistent with autosomal and X-linked recessive models were searched after filtering ‘common’ variants fulfilling the following two criteria: (1) variants with >1% minor allele frequencies in dbSNP 137, and (2) variants observed in more than six of 408 control exomes. This identified four compound heterozygous variants and three hemizygous variants. We focused on variants in PNKP, PCDH15 and ALG13 because these were predicted to be pathogenic (Table 1). ALG13 (NM_001099922.2) mutation (c.880C>T, p.Pro294Ser) is known to occur de novo only in female patients with epileptic encephalopathies,7, 8 suggesting male lethality of hemizygous mutations. Therefore, the hemizygyous ALG13 variant in the patient and its heterozygous counterpart in his unaffected mother are unlikely to be pathogenic.
The compound heterozygous PNKP (NM_007254.3) mutations (c.163G>T, p.Ala55Ser and c.874G>A, p.Gly292Arg) were of particular interest because PNKP mutations cause MCSZ syndrome or early infantile epileptic encephalopathy.2, 3, 9 The PNKP enzyme has both DNA 3′-phosphatase and DNA 5′-kinase activity and plays a key role in single- and double-stranded break DNA repair.10, 11 The altered amino residues in the patient, Ala55 and Gly292, are located within the fork head-associated and DNA phosphatase domains, respectively, and are highly conserved in vertebrates (Figure 1c). The fork head-associated domain interacts with XRCC1 and XRCC4 and has a pivotal role in DNA repair,12, 13 while the DNA phosphatase domain catalyzes the processing 3′-phosphate DNA termini to 3′-hydroxyl termini.14 Mutations in this latter domain reduce the level of PNKP protein and impair its 3′-phosphatase activity.15 Although the mutational impact to PNKP function was not investigated in our case, it is very likely that PNKP mutations cause MCSZ in the patient, who showed typical clinical features (Table 2).
The other compound heterozygous mutations (c.195G>A, p.Met65Ile, and c.1210A>C, p.Ser404Arg) in PCDH15 (NM_001142763.1) could be associated with some clinical features of our patient. PCDH15 encodes a member of the cadherin superfamily mediating calcium-dependent cell–cell adhesion. Its mutations are causative of Usher syndrome type 1F (OMIM #602083),16, 17 which is an autosomal recessive disease characterized by retinitis pigmentosa with congenital sensorineural hearing impairment.18 Our patient presented with bilateral hearing impairment from birth, suggesting PCDH15 mutations caused sensorineural hearing loss without ophthalmologic features. However, because the onset of retinitis pigmentosa in Usher syndrome is around the prepubertal period, careful follow-up for ophthalmologic symptoms is required for this patient.
In conclusion, whole-exome sequencing successfully identified causative mutations in two genes that complementarily explain the complete clinical features of the patient.
References
McKinnon, P. J. DNA repair deficiency and neurological disease. Nat. Rev. Neurosci. 10, 100–112 (2009).
Shen, J., Gilmore, E. C., Marshall, C. A., Haddadin, M., Reynolds, J. J., Eyaid, W. et al. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat. Genet. 42, 245–249 (2010).
Poulton, C., Oegema, R., Heijsman, D., Hoogeboom, J., Schot, R., Stroink, H. et al. Progressive cerebellar atrophy and polyneuropathy: expanding the spectrum of PNKP mutations. Neurogenetics 14, 43–51 (2013).
Saitsu, H., Nishimura, T., Muramatsu, K., Kodera, H., Kumada, S., Sugai, K. et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449, 449e1 (2013).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Schwarz, J. M., Rodelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 7, 575–576 (2010).
Timal, S., Hoischen, A., Lehle, L., Adamowicz, M., Huijben, K., Sykut-Cegielska, J. et al. Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum. Mol. Genet. 21, 4151–4161 (2012).
Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E. et al. De novo mutations in epileptic encephalopathies. Nature 501, 217–221 (2013).
Carvill, G. L., Heavin, S. B., Yendle, S. C., McMahon, J. M., O'Roak, B. J., Cook, J. et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 45, 825–830 (2013).
Weinfeld, M., Mani, R. S., Abdou, I., Aceytuno, R. D. & Glover, J. N. Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem. Sci. 36, 262–271 (2011).
Caldecott, K. W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631 (2008).
Koch, C. A., Agyei, R., Galicia, S., Metalnikov, P., O'Donnell, P., Starostine, A. et al. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 23, 3874–3885 (2004).
Loizou, J. I., El-Khamisy, S. F., Zlatanou, A., Moore, D. J., Chan, D. W., Qin, J. et al. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell 117, 17–28 (2004).
Jilani, A., Ramotar, D., Slack, C., Ong, C., Yang, X. M., Scherer, S. W. et al. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3'-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J. Biol. Chem. 274, 24176–24186 (1999).
Reynolds, J. J., Walker, A. K., Gilmore, E. C., Walsh, C. A. & Caldecott, K. W. Impact of PNKP mutations associated with microcephaly, seizures and developmental delay on enzyme activity and DNA strand break repair. Nucleic Acids Res. 40, 6608–6619 (2012).
Alagramam, K. N., Yuan, H., Kuehn, M. H., Murcia, C. L., Wayne, S., Srisailpathy, C. R. et al. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum. Mol. Genet. 10, 1709–1718 (2001).
Ahmed, Z. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z., Khan, S., Griffith, A. J. et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am. J. Hum. Genet. 69, 25–34 (2001).
Yan, D. & Liu, X. Z. Genetics and pathological mechanisms of Usher syndrome. J. Hum. Genet. 55, 327–335 (2010).
Acknowledgements
We would like to thank the patient’s family for participating in this work. We also thank Nobuko Watanabe for her technical assistance. This study was supported by the Ministry of Health, Labour and Welfare of Japan, a Grant-in-Aid for Scientific Research (A), (B), and (C) from the Japan Society for the Promotion of Science, the Takeda Science Foundation, the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems, the Strategic Research Program for Brain Sciences, and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Nakashima, M., Takano, K., Osaka, H. et al. Causative novel PNKP mutations and concomitant PCDH15 mutations in a patient with microcephaly with early-onset seizures and developmental delay syndrome and hearing loss. J Hum Genet 59, 471–474 (2014). https://doi.org/10.1038/jhg.2014.51
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.51
This article is cited by
-
Mutations of the DNA repair gene PNKP in a patient with microcephaly, seizures, and developmental delay (MCSZ) presenting with a high-grade brain tumor
Scientific Reports (2022)
-
The lipogenic LXR-SREBF1 signaling pathway controls cancer cell DNA repair and apoptosis and is a vulnerable point of malignant tumors for cancer therapy
Cell Death & Differentiation (2020)
