
Abstract
Rett syndrome (RTT) is a neurodevelopmental disorder mostly caused by MECP2 mutations. We identified a de novo WDR45 mutation, which caused a subtype of neurodegeneration with brain iron accumulation, in a patient showing clinically typical RTT. The mutation (c.830+1G>A) led to aberrant splicing in lymphoblastoid cells. Sequential brain magnetic resonance imaging demonstrated that iron deposition in the globus pallidus and the substantia nigra was observed as early as at 11 years of age. Because the patient showed four of the main RTT diagnostic criteria, WDR45 should be investigated in patients with RTT without MECP2 mutations.
Similar content being viewed by others


Introduction
Rett syndrome (RTT) is a neurodevelopmental disorder characterized by regression, loss of acquired purposeful hand skill and language, gait abnormalities and stereotypic hand movements.1 In typical RTT, MECP2 mutations can be found in 95–97% of cases, and CDKL5 and FOXG1 mutations have been found in atypical RTT.1 Recently, mutations in WDR45, which plays an important role in autophagy, have been identified in a novel subtype of neurodegeneration with brain iron accumulation, called β-propeller protein-associated neurodegeneration (BPAN), which is formerly designated as static encephalopathy of childhood with neurodegeneration in adulthood.2, 3, 4 BPAN shows an unprogressive course during childhood, sudden-onset severe dystonia-parkinsonism and progressive dementia in adulthood. Characteristic brain magnetic resonance imaging findings include iron deposition in the globus pallidus and substantia nigra, and hyperintensity of the substantia nigra with a central band of hypointensity in T1-weighted images.3 Interestingly, 7 of 23 patients with a WDR45 mutation showed Rett-like features, suggesting a possible involvement of WDR45 mutations in RTT.5 Here we report a patient with typical RTT possessing a de novo WDR45 mutation.
Materials and methods
A 14-year-old Japanese girl was born to non-consanguineous parents as a first child after an uneventful pregnancy. There was no familial history of neurological diseases. Although her initial development was normal as she controlled her head at 4 months of age, developmental milestones were gradually delayed and she learned to walk at 1 year and 7 months. During infancy, she played with toys and started to talk at ∼12 months of age. Her walking developed repetitive atonic episodes and an electroencephalogram showed focal irregular polyspikes and waves, and hence she was diagnosed with epilepsy. She was administered antiepileptic agents and carbamazepine was found to be effective. She gradually lost hand function and verbal communications by 4 years of age, and developed stereotypic hand movements such as continuous rubbing and licking, and dystonia. Although she now walks alone with an ataxic gait, her hand skills have regressed and she cannot talk. She shows hyperventilation, abnormally deep breathing, bruxism during waking periods, hypotonia, peripheral vasomotor disturbance, kyphosis, small cold hands and feet, sudden inappropriate laughing, diminished response to pain and intense eye communication. Sleep disturbance and microcephaly were unobserved. Brain magnetic resonance imaging at 3 and 4 years of age showed no remarkable findings (Figures 1a–d). However, T2-weighted images (WI) revealed mild hypointensity in the globus pallidus and the substantia nigra at 11 years of age (Figures 1e and f). At 14 years, this T2 shortening progressed (Figures 1g and h), and this hypointensity was obvious in T2*WI (Figures 2c and d). T1WI showed hyperintensity of the substantia nigra with a weak central band of T1WI hypointensity (Figure 2b). Experimental protocols for genetic analysis were approved by the Institutional Review Board of Yokohama City University School of Medicine. Clinical information and peripheral blood samples were acquired from family members after obtaining written informed consent.

T2-weighted axial images of the patient with a WDR45 mutation. (a–h) T2-weighted axial images of the patient. ((a, b) at 3 years and 6 months; (c, d) at 4 years and 4 months; (e, f) at 11 years and 8 months; (g, h) at 14 years and 6 months) Brain magnetic resonance imaging (MRI) at 3 and 4 years revealed no remarkable findings in the brain structure, volume and signal intensity (a–d). Mild T2 hypointensity in the globus pallidus (e, arrow) and the substantia nigra (f, arrowhead) was noticed at 11 years of age. At 14 years, T2WI showed strong hypointensity in the globus pallidus (g, arrow) and the substantia nigra (h, arrowhead). mo, months; yr, years.

Brain magnetic resonance imaging (MRI) of the patient at 14 years of age. T1-weighted (a, b), and T2*-weighted (c, d) axial images of the patient at 14 years and 6 months of age. T1-weighted images show normal findings in the globus pallidus (a), but hyperintensity of the substantia nigra with a weak central band of T1WI hypointensity (b, arrowhead). T2*-weighted images show strong hypointensity in the substantia nigra (c, arrowhead) and the globus pallidus (d, arrow).
Sanger sequencing
MECP2 mutation was sequenced by Sanger method. Parental samples were also sequenced with respect to identified variants.
Whole exome sequencing
Genomic DNA was isolated from peripheral blood leukocytes using QuickGene 610L (Wako, Osaka, Japan), captured using the SureSelect Human All Exon v4 Kit (51 Mb; Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina HiSeq2000 (Illumina, San Diego, CA, USA) with 101 bp paired-end reads. Four samples were run in one lane of the flow cell. Exome data processing, variant calling and variant annotation were performed as previously described.4 The WDR45 mutation was confirmed by Sanger sequencing.
Reverse transcriptase-PCR
Lymphoblastoid cell lines were established from the patient. Reverse transcriptase-PCR using total RNA extracted from lymphoblastoid cell lines was performed as previously described.6 Briefly, total RNA was extracted using the RNeasy Plus Mini kit (Qiagen, Tokyo, Japan), and 4 μg subjected to reverse transcription. For PCR, 2 μl of complementary DNA was used with primer ex8-9-F spanning exons 8 and 9 (5′-GTGGACCTGGCGAGCACAAAG-3′) and primer ex11-12-R spanning exons 11 and 12 (5′-AACTCTGTCATTGCCATCTGCGTAG-3′). The PCR reaction consisted of five cycles of 98 °C for 10 s, 72 °C for 30 s, five cycles of 98 °C for 10 s, 70 °C for 30 s, five cycles of 98 °C for 10 s, 68 °C for 30 s and 30 cycles of 98 °C for 10 s, 66 °C for 30 s. PCR products were electrophoresed on a 10% polyacrylamide gel and sequenced. PCR products were purified using the QIAEXII Gel extraction kit (Qiagen).
Results and Discussion
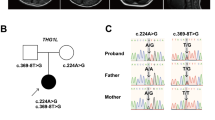
We detected c.602C>T (p.Sla201Val) mutation of MECP2 in the patient by Sanger sequencing. However, we consider this mutation was not pathogenic because the mutation was inherited from her healthy mother. The mutation was also found in 3 males and 8 females of our 574 in-house control exomes (281 males and 293 females). By whole exome sequencing, we identified a splice donor site mutation (c.830+1G>A) in the WDR45 gene, which is absent from the proband’s parents, indicating that the mutation occurred de novo (Figure 3a). The mutation was absent in the 6500 exomes sequenced by the National Heart, Lung, and Blood Institute exome project and our 574 in-house control exomes. Reverse transcriptase-PCR and sequencing revealed aberrant splicing in which 82 bp intronic sequences were retained by the use of a cryptic splice donor site within intron 10, generating a premature stop codon (p.Leu278*) (Figures 3b and c).

Mutation leading to aberrant splicing in the patient’s lymphoblastoid cell lines. (a) Location of the WDR45 mutation. A splice donor site mutation (c.830+1G>A) in the patient, which was absent from her parents, was confirmed by Sanger sequencing. (b) Reverse transcriptase (RT)-PCR analysis using lymphoblastoid cell lines derived from the patient and a control. A single band (472 bp), corresponding to the wild-type allele, was amplified using a control complementary DNA (cDNA) template. A longer aberrant band was detected from the patient’s cDNA. (c) Schematic representation of the wild-type and mutant transcripts determined by sequencing PCR products, and primers used for the analysis. The upper band (551 bp) has a 82-bp insertion of the entire intron 10 sequence, leading to a frameshift. A full color version of this figure is available at the Journal of Human Genetics journal online.
Although the brain magnetic resonance imaging at 11 years of age showed iron deposition in the globus pallidus and the substantia nigra, clinical features showed no aggravation at this time. Thus, there is no correlation between iron deposition and clinical phenotype. The fact that iron deposition preceded neurological decline is important information for elucidating BPAN pathogenesis that is caused by autophagy impairment.
The patient showed regression and stabilization, and fulfilled four of the main revised RTT diagnostic criteria,1 indicating that she clinically showed typical RTT. In autopsied RTT brains, tyrosine hydroxylase activity is reduced in the substantia nigra, and may cause hypofunction of the nigrostriatal dopamine neurons involved in modulating posture and locomotion.7 Dystonia and parkinsonism are also seen in older RTT patients as well as BPAN.8 Therefore, substantia nigra dysfunction might be involved in both RTT and BPAN, facilitating our understanding of the pathomechanism of RTT caused by MECP2 mutations. We recommend that WDR45 should be checked in RTT patients without MECP2 mutation.
References
Neul, JL, Kaufmann, WE, Glaze, DG, Christodoulou, J, Clarke, AJ, Bahi-Buisson, N et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann. Neurol. 68, 944–950 (2010).
Haack, TB, Hogarth, P, Kruer, MC, Gregory, A, Wieland, T, Schwarzmayr, T et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 91, 1144–1149 (2012).
Kruer, MC, Boddaert, N, Schneider, SA, Houlden, H, Bhatia, KP, Gregory, A et al. Neuroimaging features of neurodegeneration with brain iron accumulation. AJNR Am. J. Neuroradiol. 33, 407–414 (2012).
Saitsu, H, Nishimura, T, Muramatsu, K, Kodera, H, Kumada, S, Sugai, K et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449 449e441 (2013).
Hayflick, SJ, Kruer, MC, Gregory, A, Haack, TB, Kurian, MA, Houlden, HH et al. beta-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain 136, 1708–1717 (2013).
Saitsu, H, Kato, M, Mizuguchi, T, Hamada, K, Osaka, H, Tohyama, J et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet. 40, 782–788 (2008).
Segawa, M Early motor disturbances in Rett syndrome and its pathophysiological importance. Brain. Dev. 27 (Suppl 1), S54–S58 (2005).
FitzGerald, PM, Jankovic, J, Glaze, DG, Schultz, R & Percy, AK Extrapyramidal involvement in Rett’s syndrome. Neurology 40, 293–295 (1990).
Acknowledgements
We thank the patient’s family for their participation in this study. We also thank Nobuko Watanabe for her technical assistance. This work was supported by the Ministry of Health, Labour, and Welfare of Japan; the Japan Society for the Promotion of Science (a Grant-in-Aid for Scientific Research (B) from (25293085, 25293235), a Grant-in-Aid for Scientific Research (A) (13313587)); the Takeda Science Foundation; the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems; the Strategic Research Program for Brain Sciences (11105137); and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (12024421).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ohba, C., Nabatame, S., Iijima, Y. et al. De novo WDR45 mutation in a patient showing clinically Rett syndrome with childhood iron deposition in brain. J Hum Genet 59, 292–295 (2014). https://doi.org/10.1038/jhg.2014.18
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.18
Keywords
This article is cited by
-
Correlation of dystonia severity and iron accumulation in Rett syndrome
Scientific Reports (2021)
-
Monogenic disorders that mimic the phenotype of Rett syndrome
neurogenetics (2018)
-
RNA sequencing and proteomics approaches reveal novel deficits in the cortex of Mecp2-deficient mice, a model for Rett syndrome
Molecular Autism (2017)
-
WDR45 mutations in three male patients with West syndrome
Journal of Human Genetics (2016)
-
Early-onset epileptic encephalopathy as the initial clinical presentation of WDR45 deletion in a male patient
European Journal of Human Genetics (2016)
