
Abstract
Aminoacylation is the process of attaching amino acids to their cognate tRNA, and thus is essential for the translation of mRNA into protein. This direct interaction of tRNA with amino acids is catalyzed by aminoacyl-tRNA synthetases. Using whole-exome sequencing, we identified compound heterozygous mutations [c.169T>C (p.Tyr57His) and c.1485dup (p.Lys496*)] in QARS, which encodes glutaminyl-tRNA synthetase, in two siblings with early-onset epileptic encephalopathy (EOEE). Recessive mutations in QARS, including the loss-of-function missense mutation p.Tyr57His, have been reported to cause intractable seizures with progressive microcephaly. The p.Lys496* mutation is novel and causes truncation of the QARS protein, leading to a deletion of part of the catalytic domain and the entire anticodon-binding domain. Transient expression of the p.Lys496* mutant in neuroblastoma 2A cells revealed diminished and aberrantly aggregated expression, indicating the loss-of-function nature of this mutant. Together with the previous report, our data suggest that abnormal aminoacylation is one of the underlying pathologies of EOEE.
Similar content being viewed by others

Introduction
Aminoacylation is the process of attaching amino acids to their cognate tRNA. This direct interaction is catalyzed by the aminoacyl-tRNA synthetase (ARS) family of enzymes.1 In humans, 37 nuclear genes encode ARS enzymes, and to date, 22 of 37 ARS genes were shown to be mutated in human diseases.2, 3, 4, 5, 6
QARS encodes glutaminyl-tRNA synthetase. Two families with autosomal recessive primary microcephaly and intractable seizures harboring compound heterozygous QARS mutations have been recently reported.7 In this study, we present the detailed molecular and clinical analysis of a Japanese family with two siblings affected with early-onset epileptic encephalopathies (EOEEs) in which whole-exome sequencing confirmed the presence of QARS mutations.
Materials and Methods
Subjects
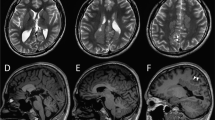
We investigated two affected siblings (II-1 and II-3) and their parents and unaffected two siblings. Clinical information and peripheral blood samples were obtained from family members after obtaining their written informed consent. A summary of the clinical features of two patients along with four patients previously reported7 is presented in Table 1, and their case reports are shown in Supplementary Information. Both patients developed intractable seizures during the neonatal period, and showed severe developmental delays, involuntary movements and intellectual disabilities. Electroencephalogram demonstrated epileptic activities, and also temporarily showed hypsarrhythmia or suppression-burst patterns (Figures 1a and b). Brain magnetic resonance imaging revealed progressive cerebral atrophy with white matter volume loss, myelination delay, ventriculomegaly and a thin corpus callosum in both individuals (Figures 1c–f). They exhibited signs of excitement, such as crying, bending backward, excessive sweating, hypertonicity and sleep disturbance. These occurred from 2.5 to 6 years of age in individual II-1, and from 5 to 10 years of age (at present day) in individual II-3. Both individuals also experienced a variety of seizures, mainly partial seizures, which worsened with fever. At birth, their head circumferences were normal (II-1: 32.0 cm (−0.9 s.d.s); II-3: 33.0 cm (−0.2 s.d.s)), but acquired microcephaly occurred in both: the head circumference of II-1 was 44 cm (−3.2 s.d.) at 2 years and 8 months, whereas that of II-3 was 35 cm (−2.9 s.d.) at 3 months. This study was approved by the Institutional Review Board of the Yokohama City University School of Medicine.

Electroencephalogram (EEG) and brain magnetic resonance imaging (MRI) of individuals with QARS mutations. (a) The EEG of II-1 showed hypsarrhythmia at 10 months. (b) The EEG of II-3 showed multifocal sharp, spike and spike-and-wave complexes at 6 days of age. (c–f) T2-weighted axial (c, e) and sagittal (d, f) images of II-1 (c, d) and II-3 (e, f). Diffuse cerebral atrophy with severe white matter volume loss, hypomyelination, hypoplastic corpus callosum, enlargement of the lateral ventricles and cerebellar atrophy were observed in both individuals. II-1 showed T2 elongation in the occipital lobes (arrows).
Results and Discussion
We conducted whole-exome sequencing to identify the genetic cause(s) of the two patients (Figure 2a). Genomic DNA was captured using the SureSelect Human All Exon Kit v5 (Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina HiSeq2000 (Illumina, San Diego, CA, USA) with 101 bp pair-end reads. Exome data processing, variant calling and variant annotation were performed as previously described.8

Familial pedigree, QARS mutations and their protein localization. (a) Familial pedigree and QARS mutations in familial members. (b) Schematic QARS proteins with InterPro domains (http://www.ebi.ac.uk/interpro/). Locations of each mutation are depicted. Previously reported mutations are shown in black and mutations identified in this study in red. (c) Localization of AcGFP1-tagged QARS proteins in neuroblastoma 2A cells. AcGFP1-tagged wild-type (WT) QARS (top panels, green) and the p.Tyr57His mutant (middle panels, green) were localized in the cytosolic compartment. Expression of the p.Lys496* mutant (bottom panels, green) was severely diminished or aggregated. The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). Scale bars, 10 μm.
The average mean read depth of the protein-coding regions in RefSeq ranged from 133 to 136, and approximately 96.7% of target coding sequences were covered by 10 or more reads (Supplementary Table 1). We filtered out common variants in dbSNP135, variants in dbSNP137NonFlagged data (‘clinically associated’ variants are excluded in this data) and variants found in more than 2 of our 575 in-house control exomes. As a result, we identified 152 and 127 rare protein-altering and splice site variants in individuals II-1 and II-3, respectively (Supplementary Table 1).
Based on an autosomal or X-linked recessive model, we focused on homozygous or compound heterozygous autosomal variants, and hemizygous variants on the X chromosome that were commonly seen in the two siblings. This analysis revealed only one autosomal candidate gene, QARS (NM_005051.2; Supplementary Table 2). Sanger sequencing confirmed that a novel QARS nonsense mutation, c.1485dup (p.Lys496*), was inherited from their father, and a missense mutation, c.169T>C (p.Tyr57His), was inherited from their mother (Figure 2a). Two other unaffected siblings only carried the c.1485dup (p.Lys496*) mutation. Neither mutation was found in the 6500 exomes of the National Heart, Lung and Blood Institute Exome Sequencing Project Variant Server, whereas c.169T>C was not found in any of our in-house 575 Japanese control exomes, and c.1485dup was only found in one of these control exomes.
Four online protein prediction programs (SIFT, Polyphen-2, PhyloP and MutationTaster; URLs listed in Supplementary Information) predicted the p.Tyr57His mutation to be damaging (Supplementary Table 2). The p.Tyr57His mutation occurs at an amino acid that is highly evolutionarily conserved among mammals, vertebrates, flies, nematodes and plants (Figure 2b). In addition, the p.Tyr57His mutation found in our family was previously identified in another family with autosomal recessive primary microcephaly.7 The p.Tyr57His mutation was demonstrated to decrease QARS aminoacylation activity to less than 10% that of the wild-type protein, indicating that it is a loss-of-function mutant.7 On the other hand, the p.Lys496* is predicted to cause truncation of QARS, involving loss of part of the catalytic domain and the entire anticodon-binding domain, registered as IPR020058 and IPR020059 in InterPro, respectively, suggesting that the function of the mutant protein would be severely impaired (Figure 2b). To examine the effect of the two QARS mutations, we performed transient expression experiments in neuroblastoma 2A cells. The p.Tyr57His mutant showed a similar expression pattern to wild type; however, expression of the p.Lys496* mutant was severely diminished or aggregated in most of the cells (Figure 2c). In addition, the c.1485dup mutant transcript possessing a premature stop codon is considered to undergo nonsense-mediated decay in patient cells. Therefore, it is very likely that the c.1485dup mutation would lead to loss-of-function of QARS by diminished expression both at mRNA and protein levels.
The clinical features of our two siblings are consistent with those of previous cases, including intractable seizures starting in the early neonatal period, postnatal microcephaly, cerebral atrophy with reduced white matter, hypomyelination, severe developmental delay and intellectual disability. Notably, our cases showed excited states similar to individual I-1 and -2 in Zhang’s report.7 These episodes associated with excitement may be a key feature for suspecting QARS mutations. In addition, seizures and hypomyelination are most commonly observed in disorders with abnormality of ARS genes,4, 6, 7, 9, 10, 11, 12, 13, 14 Therefore, abnormal aminoacylation is likely to be an underlying cause in EOEE, especially when hypomyelination and/or white matter abnormalities were seen.
In conclusion, we have described two siblings with EOEE who harbor compound heterozygous mutations in QARS. Further genetic analysis of the aminoacylation process is needed to obtain a comprehensive causal view of epileptic encephalopathy.
The following supporting information is available for this article:
Supplementary case reports, and Materials and Methods.
References
Ibba, M . & Soll, D . Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 69, 617–650 (2000).
Abbott, J. A ., Francklyn, C. S . & Robey-Bond, S. M . Transfer RNA and human disease. Front. Genet. 5, 158 (2014).
Konovalova, S . & Tyynismaa, H . Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol. Genet. Metab. 108, 206–211 (2013).
Wolf, N. I ., Salomons, G. S ., Rodenburg, R. J ., Pouwels, P. J ., Schieving, J. H . & Derks, T. G . et al. Mutations in RARS cause hypomyelination. Ann. Neurol. 76, 134–139 (2014).
Casey, J. P ., McGettigan, P ., Lynam-Lennon, N ., McDermott, M ., Regan, R . & Conroy, J . et al. Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol. Genet. Metab. 106, 351–358 (2012).
Sofou, K ., Kollberg, G ., Holmström, M ., Dávila, M ., Darin, N . & Gustafsson, C. M . et al. Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome. Mol. Genet. Genomic Med. (e-pub ahead of print 23 October 2014; doi:10.1002/mgg3.115).
Zhang, X ., Ling, J ., Barcia, G ., Jing, L ., Wu, J . & Barry, B. J . et al. Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am. J. Hum. Genet. 94, 547–558 (2014).
Saitsu, H ., Nishimura, T ., Muramatsu, K ., Kodera, H ., Kumada, S . & Sugai, K . et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449 449e441 (2013).
Taft, R. J ., Vanderver, A ., Leventer, R. J ., Damiani, S. A ., Simons, C . & Grimmond, S. M . et al. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet. 92, 774–780 (2013).
Scheper, G. C ., van der Klok, T ., van Andel, R. J ., van Berkel, C. G ., Sissler, M . & Smet, J . et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 39, 534–539 (2007).
Steenweg, M. E ., Ghezzi, D ., Haack, T ., Abbink, T. E ., Martinelli, D . & van Berkel, C. G . et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate 'LTBL' caused by EARS2 mutations. Brain 135, 1387–1394 (2012).
Bayat, V ., Thiffault, I ., Jaiswal, M ., Tetreault, M ., Donti, T . & Sasarman, F . et al. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 10, e1001288 (2012).
Elo, J. M ., Yadavalli, S. S ., Euro, L ., Isohanni, P ., Gotz, A . & Carroll, C. J . et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum. Mol. Genet 21, 4521–4529 (2012).
Edvardson, S ., Shaag, A ., Kolesnikova, O ., Gomori, J. M ., Tarassov, I . & Einbinder, T . et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 81, 857–862 (2007).
Acknowledgements
We thank the patients and their families for their participation in this study. We also thank Nobuko Watanabe for her excellent technical assistance. This work was supported by a Grant-in-Aid for Young Scientists (B) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (26860816) and the Ministry of Health, Labour and Welfare of Japan; Grants-in-Aid for Scientific Research B (25293085, 25293235) and A (13313587), and challenging Exploratory Research (26670505) from the Japan Society for the Promotion of Science; the Takeda Science Foundation; the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency; the Strategic Research Program for Brain Sciences (11105137); and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (12024421).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors decleare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Kodera, H., Osaka, H., Iai, M. et al. Mutations in the glutaminyl-tRNA synthetase gene cause early-onset epileptic encephalopathy. J Hum Genet 60, 97–101 (2015). https://doi.org/10.1038/jhg.2014.103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.103
This article is cited by
-
Role of Mutations of Mitochondrial Aminoacyl-tRNA Synthetases Genes on Epileptogenesis
Molecular Neurobiology (2023)
-
Distinct pathogenic mechanisms of various RARS1 mutations in Pelizaeus-Merzbacher-like disease
Science China Life Sciences (2021)
-
Severe growth deficiency, microcephaly, intellectual disability, and characteristic facial features are due to a homozygous QARS mutation
neurogenetics (2017)
-
Aminoacyl-tRNA synthetase dependent angiogenesis revealed by a bioengineered macrolide inhibitor
Scientific Reports (2015)
