
Abstract
Charcot–Marie–Tooth disease (CMT) is a genetically and clinically heterogeneous hereditary motor and sensory neuropathy signified by a distal symmetric polyneuropathy. The most frequent subtype is type 1A (CMT1A) caused by duplication in chromosome 17p12 that includes PMP22. This study reports a woman with a family history of CMT1A due to PMP22 duplication. However, she presented with a more severe phenotype than her sibling or ancestors and was found to have a PMP22 triplication instead of the duplication. This was caused by de novo mutation on her affected mother’s duplication chromosome. Her lower limb magnetic resonance imaging revealed severe diffused atrophy and fatty replacement. However, her affected sister with typical PMP22 duplication showed almost intact lower limb. Triplication patient’s median motor nerve conduction velocity was far lower compared with her sister. Her onset age was faster (8 years) than her sister (42 years). CMT1A triplication might be generated by a female-specific chromosomal rearrangement mechanism that is different from the frequent paternal-originated CMT1A duplication. It also suggests that the wide phenotypic variation of CMT1A might be partly caused by unstable genomic rearrangement, including PMP22 triplication.
Similar content being viewed by others


Introduction
Charcot–Marie–Tooth disease (CMT) is a group of genetically and clinically heterogeneous peripheral nerve disorders signified by a distal symmetric polyneuropathy. More than 70 genes have been reported as the underlying causes of CMT.1 Particularly, duplication and deletion of PMP22 at chromosome 17p12 cause CMT type 1A (CMT1A, MIM #118220)2 and hereditary neuropathy with liability to pressure palsies (HNPP, MIM #162500), respectively.3 PMP22 duplication and deletion are the most frequent genetic cause of peripheral neuropathies.4, 5, 6 Major genetic mechanism of CMT1A and hereditary neuropathy with liability to pressure palsies is based on recurrent nonallelelic homologous recombination. However, replication-based nonrecurrent rearrangement has been also reported in CMT1A.7, 8 Recently, Liu et al.9 reported severe phenotype of CMT1A with PMP22 triplication.
Wide phenotypic spectrum has been reported in CMT1A patients.10, 11 Here, we identified a CMT1A family with wide phenotypic difference. Subsequent genetic study identified that a patient with severe clinical symptoms had unusual PMP22 triplication.
Materials and methods
Subjects and clinical examination
This study examined a Korean CMT1A family of five affected individuals (Figure 1a). Clinical information including motor and sensory impairments and deep tendon reflexes was obtained in a standardized manner. Magnetic resonance imaging study was performed on the lumbar spine and lower limb including thigh and calf of the patients at a supine position using a 1.5-T system (Siemens Vision, Erlangen, Germany). Physical disability was determined by two scales: functional disability scale12 and CMT neuropathy score.13 All participants provided written informed consents. This study was approved by the Institutional Review Board for Sungkyunkwan University School of Medicine, Samsung Medical Center.

A Charcot–Marie–Tooth disease type 1A (CMT1A) family with PMP22 duplication and triplication. (a) Pedigree. Gray and black symbols present affected individuals with PMP22 duplication and triplication, respectively. Open symbols indicates unaffected individuals. Proband is indicated by an arrow. Haplotypes of eight microsatellites are indicated at the bottom of all examined individuals. (b) Quantification of PMP22 dosage by real-time PCR. When PMP22 dosage of healthy control was expressed as 1.0, proband (III-2) showed about two folds dosage, whereas other affected individuals showed around 1.5-folds (CTL(N): unaffected control, CTL(+): CMT1A with PMP22 duplication and CTL(−): hereditary neuropathy with liability to pressure palsies (HNPP) with PMP22 deletion). (c) Microsatellite genotyping. Microsatellites within 17p12 were amplified using FAM- or HEX-labeled primers and resolved using an automatic sequencer ABI 3130XL. Figure shows chromatograms of D17S4A. Control (normal copy number) shows two peaks, but affected samples shows three peaks. In proband (III-2), however, the largest allele particularly shows the highest peak, suggesting dual dosage. A full color version of this figure is available at the Journal of Human Genetics journal online.
Molecular studies
Genomic DNA was isolated from blood by using a QIAamp DNA purification kit (Qiagen, Hilden, Germany). Copy number of PMP22 (17p12) was determined by two methods: genotyping of microsatellites within the duplication region and quantification of PMP22 dosage by real-time PCR.8 Eight microsatellites were amplified for genotyping with a hexaplex PCR (D17S921, D17S9A, D17S9B, D17S918, D17S4A and D17S2230) or single PCR (D17S1296 and D17S1357) using FAM- or HEX-labeled primers. Real-time PCR for PMP22 was performed by using a SYBR Green Supermix and CFX96 PCR system (Bio-Rad, Hercules, CA, USA). Mutations for more than 70 CMT-related genes were determined by targeted sequencing of coding exons using a custom-ordered platform and a HiSeq2000 Genome Analyzer (Illumina, San Diego, CA, USA).
Results
Identification of CMT1A triplication
DNA test revealed an unusual PMP22 triplication in a severely affected woman (Figure 1a, III-2). All other mildly affected family members (II-2, II-3, III-1 and III-4) showed typical PMP22 duplication. Real-time PCR showed high PMP22 dosage (approximately two-fold compared with control) for triplication patient (III-2) compared with other affected individuals (approximately 1.5-fold compared with control) (Figure 1b). Haplotype analysis of eight microsatellites suggested that the triplication was caused by a de novo mutation from her mother (II-2) due to intrachromosomal rearrangement between sister chromatids having PMP22 duplication, because alleles from the triplication matched with one of the duplicated alleles exactly (Figures 1a and c).
Targeted sequencing for three affected individuals (II-3, III-2 and III-4) revealed many nonsynonymous variants in the CMT-related genes. However, no variant was considered as causative because all variants were found in the controls or reported in the dbSNP141 and 1000 Genomes Database (Supplementary Table 1).
Clinical manifestations
Patient with PMP22 triplication (III-2: proband)
Patient with PMP22 triplication experienced frequent falling and noticed muscle weakness of the distal lower limbs at 8 years old. Sensory ataxia was observed at 25 years old. She walked with cane at 34 years old. Examination at 48 years of age revealed bilateral distal muscle weakness and atrophy predominantly at lower limbs. Bilateral pes cavus, steppage gait and atrophy of intrinsic foot and calf muscles were noted. Her sensitivity to pinprick, touch, position and vibration were decreased. Vibration and position senses were more severely disturbed than pain and touch senses. Her knee and ankle jerks were absent. No pyramidal or cerebellar signs were detected. Her functional disability scale and CMT neuropathy score were 4 and 27, respectively.
Patient with PMP22 duplication (III-4)
Her younger sister with PMP22 duplication of the proband noticed weakness at the distal lower limbs at 42 years old. Examination at 44 years of age revealed muscle weakness without atrophy of bilateral distal muscles. Her vibration and position senses were disturbed. Her knee and ankle jerks were decreased. Functional disability scale and CMT neuropathy score were 1 and 5, respectively.
Severe symptoms in CMT1A triplication patient
Electrophysiological studies showed striking differences between the patient with PMP22 triplication and her sister with duplication (Table 1). Median motor nerve conduction velocity indicated demyelinating neuropathy in both patients. However, it was more decreased in the triplication patient (11.3 ms−1) than in her sister (29.0–30.4 ms−1). Compound muscle action potential in median nerve was more severely impaired in the triplication patient (0.7–1.0 mV) than in her sister (13.4–14.2 mV). In addition, sensory nerve action potentials in median, ulnar and sural nerves were almost lost in the early stage of her disease. However, sensory nerve action potentials were relatively preserved in her sister.
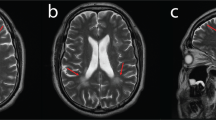
Lower limb magnetic resonance imaging including thigh and calf muscles revealed quite different condition between patients with PMP22 triplication and duplication (Figure 2). Triplication patient showed nearly all compartment muscle atrophy and fatty replacement in the calf muscles (Figures 2d–f). In addition, proximal atrophy of bilateral vastus lateralis muscles at the thigh level was observed (Figures 2a–c). But her sister with PMP22 duplication revealed almost normal muscles in the thigh and calf levels (Figures 2g–l).

Comparison of lower leg limb atrophy between PMP22 triplication (III-2, a– f) and duplication (III-4, g– l). Lower limb magnetic resonanc imagings including thigh and calf shows strikingly different muscle atrophy between two patients. Lower calf muscle magnetic resonanc imagings shows severe atrophy and fatty replacement in the whole compartments in triplication patient (d–f). At the thigh level, triplication patient also shows fatty replacement in bilateral vastus lateralis muscles (c, white arrowhead). On the contrary, all muscles were relatively intact in duplication patient (g–l).
Conclusion
This study reports a woman presented severe CMT1A with rare PMP22 triplication caused by a de novo mutation from her mother. Previous PMP22 triplication cases were also caused by de novo mutation with maternal origin.9 Moreover, all PMP22 triplication cases showed an intrachromosoal rearrangement. Usual de novo PMP22 duplication is resulted from recurrent nonallelelic homologous recombination between nonsister chromatids with the prevalence of paternal origin.14, 15, 16, 17 Therefore, de novo PMP22 triplication was suggested to be generated by a female-specific rearrangement mechanism different from the PMP22 duplication event.18
When the PMP22 triplication patient was compared with duplication patient, she exhibited far more severe symptoms with increased functional disability scale and CMT neuropathy score. Her onset was faster than her sister, and her motor nerve conduction velocity was far decreased compared with her sister. Her lower limb magnetic resonance imaging also revealed severe diffused atrophy and fatty replacement, but her sister showed almost intact. Moreover, triplication patient showed fatty replacement in the proximal leg muscles.
Wide phenotypic spectrum in CMT1A suggested that a second modifier genes or association factor(s) might be involved. Lose genotype–phenotype correlation of CMT1A might be partly caused by unstable genomic rearrangement, including PMP22 triplication.
References
Rossor,, A. M ., Polke, J. M ., Houlden, H . & Reilly, M. M . Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat. Rev. Neurol. 9, 562–571 (2013).
Lupski, J. R ., de Oca-Luna, R. M ., Slaugenhaupt, S ., Pentao, L ., Guzzetta, V . & Trask, B. J . et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66, 219–232 (1991).
Chance, P. F ., Alderson, M. K ., Leppig, K. A ., Lensch, M. W ., Matsunami, N . & Smith, B . et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 72, 143–151 (1993).
Nelis, E ., Van Broeckhoven, C ., De Jonghe, P ., Löfgren, A ., Vandenberghe, A . & Latour, P . et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur. J. Hum. Genet. 4, 25–33 (1996).
Choi, B. O ., Lee, M. S ., Shin, S. H ., Hwang, J. H ., Choi, K. G . & Kim, W. K . et al. Mutational analysis of PMP22, MPZ, GJB1, EGR2 and NEFL in Korean Charcot-Marie-Tooth neuropathy patients. Hum. Mutat. 24, 185–186 (2004).
Abe, A ., Numakura, C ., Kijima, K ., Hayashi, M ., Hashimoto, T . & Hayasaka, K . Molecular diagnosis and clinical onset of Charcot-Marie-Tooth disease in Japan. J. Hum. Genet. 56, 364–368 (2011).
Zhang, F ., Seeman, P ., Liu, P ., Weterman, M. A ., Gonzaga-Jauregui, C . & Towne, C. F . et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am. J. Hum. Genet. 86, 892–903 (2010).
Choi, B. O ., Kim, N. K ., Park, S. W ., Hyun, Y. S ., Jeon, H. J . & Hwang, J. H . et al. Inheritance of Charcot-Marie-Tooth disease 1A with rare nonrecurrent genomic rearrangement. Neurogenetics 12, 51–58 (2011).
Liu, P ., Gelowani, V ., Zhang, F ., Drory, V. E ., Ben-Shachar, S . & Roney, E . et al. Mechanism, prevalence, and more severe neuropathy phenotype of the Charcot-Marie-Tooth type 1A triplication. Am. J. Hum. Genet. 94, 462–469 (2014).
Kim, Y. H ., Chung, H. K ., Park, K. D ., Choi, K.-G ., Kim, S.-M . & Sunwoo, I.-N . et al. Comparison between clinical disabilities and electrophysiological values in Charcot–Marie–Tooth 1A patients with PMP22 duplication. J. Clin. Neurol. 8, 139–145 (2012).
Mathis, S ., Corcia, P ., Tazir, M ., Camu, W ., Magdelaine, C . & Latour, P . et al. Peripheral myelin protein 22 gene duplication with atypical presentations: a new example of the wide spectrum of Charcot-Marie-Tooth 1A disease. Neuromuscul. Disord. 24, 524–528 (2014).
Birouk, N ., Gouider, R ., Le Guern, E ., Gugenheim, M ., Tardieu, S . & Maisonobe, T . et al. Charcot-Marie-Tooth disease type 1A with 17p11.2 duplication. Clinical and electrophysiological phenotype study and factors influencing disease severity in 119 cases. Brain 120, 813–823 (1997).
Shy, M. E ., Blake, J ., Krajewski, K ., Fuerst, D. R ., Laura, M . & Hahn, A. F . et al. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology 64, 1209–1214 (2005).
Palau, F ., Löfgren, A ., De Jonghe, P ., Bort, S ., Nelis, E . & Sevilla, T . et al. Origin of the de novo duplication in Charcot-Marie-Tooth disease type 1A: unequal nonsister chromatid exchange during spermatogenesis. Hum. Mol. Genet. 2, 2031–2035 (1993).
Blair, I. P ., Nash, J ., Gordon, M. J . & Nicholson, G. A . Prevalence and origin of de novo duplications in Charcot-Marie-Tooth disease type 1A: first report of a de novo duplication with a maternal origin. Am. J. Hum. Genet. 58, 472–476 (1996).
Lopes, J ., Ravisé, N ., Vandenberghe, A ., Palau, F ., Ionasescu, V . & Mayer, M . et al. Fine mapping of de novo CMT1A and HNPP rearrangements within CMT1A-REPs evidences two distinct sex-dependent mechanisms and candidate sequences involved in recombination. Hum. Mol. Genet. 7, 141–148 (1998).
Choi, B. O ., Kim, J ., Lee, K. L ., Yu, J. S ., Hwang, J. H . & Chung, K. W . Rapid diagnosis of CMT1A duplications and HNPP deletions by multiplex microsatellite PCR. Mol. Cells 23, 39–48 (2007).
LeGuern, E ., Gouider, R ., Ravise, N ., Lopes, J ., Tardieu, S . & Gugenheim, M . et al. A de novo case of hereditary neuropathy with liability to pressure palsies (HNPP) of maternal origin: a new mechanism for deletion in 17p11.2? Hum. Mol. Genet. 5, 103–106 (1996).
Acknowledgements
This study was supported by grants of the Korean Health Technology R&D Project, Ministry of Health and Welfare (A120182) and the National Research Foundation (NRF), Ministry of Education (2014R1A2A2A01003164), Republic of Korea.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Kim, S., Lee, J., Yoon, B. et al. Severe phenotypes in a Charcot–Marie–Tooth 1A patient with PMP22 triplication. J Hum Genet 60, 103–106 (2015). https://doi.org/10.1038/jhg.2014.102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.102
