
Abstract
Several lines of evidence suggest that in Caucasian populations, mutations in the cystic fibrosis (CF) transmembrane conductance regulator (CFTR) gene are associated with susceptibility to lung disease caused by nontuberculous mycobacteria (NTM). However, there is little data available in Asian populations, in which the prevalence of CF is very low. Therefore, we investigated this potential relationship in a Korean population. Sixty patients who fulfilled the diagnostic criteria for NTM lung disease were screened for genetic alterations in the CFTR gene by whole-exon resequencing. For all identified CFTR gene variants, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) genotyping was performed. Genotype and haplotype data were compared between 360 patients with NTM lung disease and 446 healthy controls. Among 13 CFTR genetic variants that were found by whole-exon resequencing, Q1352H showed a significantly higher frequency in NTM patients than in controls, giving an odds ratio (OR) of 4.27 (95% confidence interval (CI), 1.43–12.78). A haplotype with Q1352H showed the strongest association with the disease, with an OR of 3.73 (95% CI, 1.50–9.25). Furthermore, all Q1352H alleles were associated with the V allele of the V470M variant. Our results suggest that CFTR gene variants may increase susceptibility to NTM lung disease in the Korean population. Q1352H appears to be strongly related to NTM lung disease susceptibility in the Korean population.
Similar content being viewed by others


Introduction
The incidence of lung disease caused by nontuberculous mycobacteria (NTM) in HIV-negative patients has been increasing worldwide.1, 2 A substantial proportion of these patients have no predisposing risk factors and are predominantly elderly women with characteristic radiographic findings of bronchiectasis combined with multiple small nodules.1 Mycobacterium avium complex and M. abscessus complex are known to be associated with NTM lung disease.3, 4, 5, 6, 7, 8
NTM are distributed in soil and water throughout the world. As NTM are ubiquitous, exposure to these organisms is inevitable. However, disease is rare, which suggests that normal host defense mechanisms are sufficient to prevent infection.9 Therefore, otherwise healthy individuals who develop NTM lung disease likely have specific susceptibility factors that make them vulnerable to these infections.10, 11
Cystic fibrosis (CF) is an autosomal-recessive genetic disorder and is characterized by mutations in the CFTR gene encoding the CF transmembrane conductance regulator, which result in abnormally thickened airway secretions, chronic bacterial airways infection, bronchiectasis and early death.12 NTM infections are common in patients with CF,13 and NTM isolation should lead to consideration of the diagnosis of CF in older patients, because a significant proportion of adults with NTM lung disease may have previously undiagnosed CF.14 Previous studies from some referral centers in the United States revealed that about one-third to one-half of patients with NTM lung disease had CFTR gene mutations.11, 15
CF is much more common among Caucasians than Asians.16, 17 Furthermore, the association between CFTR gene mutations and NTM lung disease has rarely been studied in non-Caucasian populations. A recent study reported that genetic alterations in the CFTR gene might be involved in susceptibility to NTM lung disease in a Japanese population.18 Our aim in the present study was to investigate whether CFTR gene variants have a role in susceptibility to NTM lung disease in a Korean population.
Materials and methods
Study population and protocol
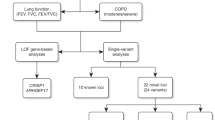
Patients who were diagnosed with NTM lung disease at Samsung Medical Center (a 1961-bed referral hospital in Seoul, Korea) between 2001 and 2010 were candidates for this study. All patients met the diagnostic criteria of the American Thoracic Society for NTM lung disease1 and had characteristic findings of NTM on high-resolution computed tomography scans, such as bilateral bronchiectasis combined with multiple small nodules and branching linear structures.19, 20 The study protocol involved three steps (Figure 1). As a first step, we performed whole-exon resequencing of CFTR gene to find disease-associated variants in 60 Korean patients with NTM lung disease. The patients were categorized based on the organism recovered at the time of diagnosis. Thirty individuals had M. avium complex (15 M. avium and 15 M. intracellulare) infections and 30 individuals had M. abscessus complex (15 M. abscessus and 15 M. massiliense) infections. For the next step, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) genotyping was performed for all identified CFTR gene variants. Blood samples from a total of 300 patients with NTM lung disease and 446 healthy controls were subjected to MALDI-TOF MS genotyping. Normal control group comprised unrelated individuals who were free of respiratory symptoms. In the final step, we compared genotype and haplotype data between 360 patients with NTM lung disease and 446 healthy controls. This study was approved by the Institutional Review Board of Samsung Medical Center (IRB approval 2008-09-016).

Steps in the study protocol: (1) whole-exon resequencing of CFTR gene was performed for the 60 patients with NTM lung disease and (2) MALDI-TOF MS genotyping was used for all identified CFTR gene variants obtained from the first step. (3) We then compared genotype and haplotype data between 360 patients with NTM lung disease and 446 healthy normal controls. CFTR, cystic fibrosis transmembrane conductance regulator; MALDI-TOF MS, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; NTM, nontuberculous mycobacteria.
Whole-exon resequencing of CFTR gene
All coding exons and flanking introns of the CFTR gene were amplified using primer sets designed by the authors (Supplementary Table S1). PCR was performed on a Thermal Cycler 9700 (Applied Biosystems, Foster City, CA, USA) as follows: 32 cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s. After treatment of the amplification product (5 μl) with shrimp alkaline phosphatase and exonuclease I (USB Corporation, Cleveland, OH, USA), sequencing was performed using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems), and the products were run on an ABI Prism 3730xl Genetic Analyzer (Applied Biosystems). The GenBank accession number of the CFTR reference sequence that we had used is NM_000492.3. To describe sequence variants, we followed the guidelines of the Human Genome Variation Society.
CFTR genotyping using MALDI-TOF MS analysis
Detection of all CFTR gene variants was carried out by high-throughput multiplex analysis on a Sequenom MassARRAY MALDI-TOF MS (Sequenom, San Diego, CA, USA). We designed specific primers flanking the mutation site and extension primers that bind adjacent to the mutation sites using MassARRAY assay design software (Supplementary Table S2). The method involves a multiplex primary PCR followed by an iPLEX reaction with a single primer of distinct mass that is specific for each genotype.
A primary PCR was performed by combining 1 μl of genomic DNA with 4 μl of PCR cocktail in the wells of a 384-well plate. The PCR cocktail comprised of 875.5 μl of water, 230.4 μl of PCR buffer, 184.3 μl of 25 mM MgCl2, 23.1 μl of dNTP mix (25 mM each dNTP), 460.8 μl of primer mix (500 nM each) and 46.1 μl of PCR enzyme. The well plate was then subjected to cycling on a thermal cycler (Model T1 plus; Biometra, Goetingen, Germany), and the cycling conditions were as follows: 1 cycle at 94 °C for 4 min, 45 cycles of 94 °C for 20 s, 56 °C for 30 s and 72 °C for 1 min, followed by a final extension step at 72 °C for 3 min. Allele discrimination reactions were carried out by adding 2 μl of iPLEX reaction mix to the dephosphorylated primary PCR reaction mix. Reactions were cycled at 94 °C for 30 s, followed by 40 cycles of 94 °C for 5 s, 52 °C for 5 s and 80 °C for 5 s, then 72 °C for 3 min. The products were spotted onto a 384-spot SpectroCHIP with a MassARRAY Nanodispenser (Sequenom). Samples were analyzed on a MassARRAY Analyzer Compact (Sequenom).
Statistical analyses
To assess the data quality of the identified genetic variants, P-values were calculated by a permutation version of the exact test for Hardy–Weinberg equilibrium.21 All data were analyzed using SAS software, version 9.3 (SAS Institute, Cary, NC, USA). To analyze genotype and allele frequency data, differences between control and disease groups were compared by a χ2 test. When an expected cell value was <5, Fisher’s exact test was used. The Bonferroni technique was used to evaluate the overall statistical significance of multiple comparisons. Haplotype estimation was based on a Bayesian algorithm using the Markov chain Monte Carlo technique.22 All P-values were based on two-sided comparisons, and P-values <0.05 were considered to indicate statistical significance.
Results
Characteristics of study population
Three hundred and sixty patients with NTM lung disease were recruited. The average age of the 360 patients was 58.5±11.8 years (range, 20.0–85.0 years). Two hundred and sixty-six patients were female and 94 were male, yielding a female-to-male ratio of 2.8:1. The NTM patients were infected as follows: M. avium complex, 69.2% (M. avium 39.4% and M. intracellulare 29.7%) and M. abscessus complex, 30.8% (M. abscessus 15.0% and M. massiliense 15.8%). The average age of the 446 controls was 51.4±5.6 years (range, 42.0–69.0 years) and the female-to-male ratio was 2.3:1.
Genetic variants of CFTR in the Korean population
As CFTR gene variants in entire coding exons and their flanking intronic regions have not been extensively evaluated in Asian populations due to the low prevalence of the CF, we undertook whole-exon resequencing of CFTR gene to find disease-associated variants in the Korean population. We detected a total of 13 CFTR genetic variants, namely, −8G>C, R31C, I125T, R297Q, V470M, S485C, I556V, E681V, T854=, S912L, P1290=, Q1352H and Q1463=. The general characteristics of the 13 variants are summarized in Table 1.
Genotype and allele frequencies in control and patient groups
Genotype distributions of CFTR gene variants were in Hardy–Weinberg equilibrium. To investigate the association between CFTR genetic variants and NTM lung disease, genotype and allele frequencies were analyzed using samples from 360 Korean patients diagnosed with NTM lung disease (2n=720) and 446 healthy controls (2n=892). The 360 patients with NTM lung disease comprised 60 patients for whom whole-exon resequencing of CFTR gene had been performed and 300 patients for whom MALDI-TOF MS genotyping results were available.
The genotype and allele frequencies at the 13 CFTR variant loci are listed in Table 2. Among the variants, Q1352H was found to be significantly associated with the NTM disease group. The genotype frequency of Q1352H was significantly higher in patients than in controls (P<0.001). The frequency of this allele was also significantly different between groups (4.9% in the NTM disease group versus 1.2% in the control group; P<0.001). As a total of 26 comparisons were made across 13 loci, the overall significance of Q1352H was revised by the Bonferroni technique. The corrected P-value was 0.0003, which is still significant. The odds ratio for an association between NTM and the CFTR Q1352H variant genotype and allele were 4.27 (95% confidence interval: 1.43–12.78) and 4.10 (95% confidence interval: 1.39–12.13), respectively. Other CFTR variants did not show any significant differences in genotype and allele frequencies between groups. Furthermore, there were no significant differences in the genotype frequencies of any of the CFTR genetic variants in patients with M. avium complex infections versus patients with M. abscessus complex infections (data not shown).
Haplotype estimations and their disease associations
Haplotype analysis was performed to gain a better understanding of the disease-associated CFTR variants using the genotype data obtained from the 806 tested samples (360 patients and 446 controls). Thirteen CFTR loci with diallelic variants were analyzed and a total of 22 haplotypes were assembled. The five haplotypes, which accounted for >95% of haplotypes, were compared with the frequency of those haplotypes in the controls (Table 3). As expected from the genotype data, the haplotype containing Q1352H showed the strongest association with NTM lung disease in our Korean population (P<0.001, corrected P=0.0003), giving an odds ratio of 3.73 (95% confidence interval: 1.50–9.25). Notably, Q1352H was observed only in the V allele background of V470M in the tested population (Table 3).
Discussion
The findings of this study support previous observations that the CFTR gene might be a susceptible gene for NTM lung disease. Notably, the Q1352H variant showed a strong association with NTM lung disease in this study, although this variant has not yet been reported in Western cohort studies of men with NTM lung disease.11, 15 The present study is the first to confirm a genetic association between CFTR gene variants and NTM lung disease in an Asian population.
The CFTR gene is located on chromosome 7q31.2 and consists of 27 exons. It encodes a transporter protein that functions as a chloride channel across cellular membranes. Mutations in the CFTR gene are known to be associated with classical CF, as well as a broad spectrum of other related diseases, including NTM lung disease. Currently, >1900 CFTR gene variants are listed in the CF mutation database (http://www.genet.sickkids.on.ca/cftr/; latest database updated on 25 April 2011). The majority of mutations have been identified in Caucasian populations, but the spectrum of mutations has not been well described in Asian populations due to the low prevalence of CF. In Korea, only 12 cases of CF have been reported in the literature thus far, of which 9 cases were confirmed by CFTR mutational analysis.17 It has been found that the mutation spectrum of the CFTR gene was highly heterogeneous and different from that seen in Caucasian populations.17 The authors of that study suggested that in Asian populations, sequencing of the entire CFTR gene might be a more rational approach than screening for a set of common CFTR mutations. Consistent with this previous study, we did not find any of the common disease-causing CFTR mutations reported in Caucasians such as F508del, G542* or N1303K.23
Of the 13 variants that we found, Q1352H was significantly associated with NTM lung disease (corrected P=0.003). The previous study in the Korean population reported that the genotype frequency of the Q1352H variant was significantly higher in patients with bronchiectasis and chronic pancreatitis than in the control group.24 Thus, the authors suggested that the Q1352H variant was strongly associated with respiratory and pancreatic disease in Koreans. This variant has also been reported in Singaporean patients with severe asthma.25 In addition, the Q1352H variant has been found in Japanese individuals with congenital bilateral absence of the vas deferens as well as in a Korean patient with alcoholic chronic pancreatitis.26, 27
It is not clear why this variant is involved in the susceptibility of chronic lung diseases and other CF-associated diseases, but the functional effect of the Q1352H variant has been reported to decrease mature CFTR protein expression by 73% and to decrease chloride current activity by 71%.24 The substitution of glutamine with histidine evokes defects in protein expression and the gating properties of the channel, because Q1352H is located in a conserved glutamine-rich sequence. Based on these findings, individuals with the Q1352H variant are expected to have slightly decreased CFTR protein and function in vivo, so that they could be susceptible to have NTM lung disease or other CF-associated diseases depending on other environmental or genetic factors.
It is of note that the average age of the 35 patients (26 female and 9 male) with the Q1352H variant was 60.6±13.0 years in our study. Twenty-three of these patients were infected with M. avium complex mycobacteria (18 M. avium and 5 M. intracellulare) and 12 with M. abscessus complex mycobacteria (2 M. abscessus and 10 M. massiliense). There were no significant differences in age, sex or NTM species between patients with the Q1352H variant and those without this variant.
The V470M variant is one of the most common polymorphisms in the CFTR gene. In-vitro studies have shown that CFTR proteins containing V470 have about half the intrinsic chloride channel activity of CFTR proteins containing M470.28 In a study of Japanese patients with M. avium complex infections, the V470 allele was observed twice as frequently as the M470 allele.18 The authors reported that the V470 allele was present mostly in patients with the IVS8 T5 variant allele; therefore, they suggested that the V470 allele may increase the penetration and severity of T5 as a disease-causing allele. In the current study, the frequency of V470 was high in both patients and controls, with a difference of 1.4-fold between the two groups. Interestingly, the Q1352H variant was only in patients with the V470 allele variant in haplotype estimation. It has been reported that the combination of Q1352H and V470 allele background of V470M results in the complete abolition of CFTR-dependent chloride transport activities.24 The haplotype of H1352 and V470 showed the strongest association with NTM lung disease in the present study, suggesting that CFTR gene variants are associated with NTM lung disease in the Korean population.
The I556V variant was first described in a French patient with moderate pulmonary symptoms and positive sweat chloride.29 I556 is highly conserved across vertebrates, similar to Q1352; thus, a change in the amino acid at these positions may cause a decrease in cell chloride current.24 Although the V556 genotype was observed at higher frequency in NTM patients than in controls, this difference was insignificant (7.5% and 4.7%, respectively; P=0.096). There was also no difference in allele frequencies between groups (4.0% and 2.4%, respectively; P=0.054). Further studies of larger cohorts of Asians are required to clarify the association between the I556V variant and NTM lung disease.
In a study of a Singaporean population, I125T was found to be associated with chronic pulmonary disease, especially idiopathic bronchiectasis.25 However, in this previous study, only six idiopathic bronchiectasis patients were examined, making it difficult to generalize the results; furthermore, the functional effect of this variant is still not known. In the present study, T125 showed a much higher allele frequency in controls than expected (0.6% and 0.9%, respectively; P=0.428). Although the difference was not significant, this finding suggests that T125 may be a rare polymorphism rather than a pathogenic allele.
Taken together, the results from our work suggest that CFTR gene variants are associated with NTM lung disease in the Korean population. Notably, the Q1352H variant appears to be strongly related with NTM lung disease.
References
Griffith, D. E., Aksamit, T., Brown-Elliott, B. A., Catanzaro, A., Daley, C., Gordin, F. et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care Med. 175, 367–416 (2007).
Daley, C. L. & Griffith, D. E. Pulmonary non-tuberculous mycobacterial infections. Int. J. Tuberc. Lung Dis. 14, 665–671 (2010).
Prevots, D. R., Shaw, P. A., Strickland, D., Jackson, L. A., Raebel, M. A., Blosky, M. A. et al. Nontuberculous mycobacterial lung disease prevalence at four integrated health care delivery systems. Am. J. Respir. Crit. Care Med. 182, 970–976 (2010).
Adjemian, J., Olivier, K. N., Seitz, A. E., Holland, S. M. & Prevots, D. R. Prevalence of nontuberculous mycobacterial lung disease in USA Medicare beneficiaries. Am. J. Respir. Crit. Care Med. 185, 881–886 (2012).
Hayashi, M., Takayanagi, N., Kanauchi, T., Miyahara, Y., Yanagisawa, T. & Sugita, Y. Prognostic factors of 634 HIV-negative patients with Mycobacterium avium complex lung disease. Am. J. Respir. Crit. Care Med. 185, 575–583 (2012).
Koh, W. J., Jeong, B. H., Jeon, K., Lee, N. Y., Lee, K. S., Woo, S. Y. et al. Clinical significance of the differentiation between Mycobacterium avium and Mycobacterium intracellulare in M. avium complex lung disease. Chest 142, 1482–1488 (2012).
Jarand, J., Levin, A., Zhang, L., Huitt, G., Mitchell, J. D. & Daley, C. L. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin. Infect. Dis. 52, 565–571 (2011).
Koh, W. J., Jeon, K., Lee, N. Y., Kim, B. J., Kook, Y. H., Lee, S. H. et al. Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am. J. Respir. Crit. Care Med. 183, 405–410 (2011).
Guide, S. V. & Holland, S. M. Host susceptibility factors in mycobacterial infection. Genetics and body morphotype. Infect. Dis. Clin. North Am. 16, 163–186 (2002).
Sexton, P. & Harrison, A. C. Susceptibility to nontuberculous mycobacterial lung disease. Eur. Respir. J. 31, 1322–1333 (2008).
Kim, R. D., Greenberg, D. E., Ehrmantraut, M. E., Guide, S. V., Ding, L., Shea, Y. et al. Pulmonary nontuberculous mycobacterial disease: prospective study of a distinct preexisting syndrome. Am. J. Respir. Crit. Care Med. 178, 1066–1074 (2008).
Lobo, J., Rojas-Balcazar, J. M. & Noone, P. G. Recent advances in cystic fibrosis. Clin. Chest Med. 33, 307–328 (2012).
Olivier, K. N., Weber, D. J., Wallace, R. J., Faiz, A. R., Lee, J. H., Zhang, Y. et al. Nontuberculous mycobacteria. I: multicenter prevalence study in cystic fibrosis. Am. J. Respir. Crit. Care Med. 167, 828–834 (2003).
Rodman, D. M., Polis, J. M., Heltshe, S. L., Sontag, M. K., Chacon, C., Rodman, R. V. et al. Late diagnosis defines a unique population of long-term survivors of cystic fibrosis. Am. J. Respir. Crit. Care Med. 171, 621–626 (2005).
Ziedalski, T. M., Kao, P. N., Henig, N. R., Jacobs, S. S. & Ruoss, S. J. Prospective analysis of cystic fibrosis transmembrane regulator mutations in adults with bronchiectasis or pulmonary nontuberculous mycobacterial infection. Chest 130, 995–1002 (2006).
Yamashiro, Y., Shimizu, T., Oguchi, S., Shioya, T., Nagata, S. & Ohtsuka, Y. The estimated incidence of cystic fibrosis in Japan. J. Pediatr. Gastroenterol. Nutr. 24, 544–547 (1997).
Jung, H., Ki, C. S., Koh, W. J., Ahn, K. M., Lee, S. I., Kim, J. H. et al. Heterogeneous spectrum of CFTR gene mutations in Korean patients with cystic fibrosis. Korean J. Lab. Med. 31, 219–224 (2011).
Mai, H. N., Hijikata, M., Inoue, Y., Suzuki, K., Sakatani, M., Okada, M. et al. Pulmonary Mycobacterium avium complex infection associated with the IVS8-T5 allele of the CFTR gene. Int. J. Tuberc. Lung Dis. 11, 808–813 (2007).
Koh, W. J., Lee, K. S., Kwon, O. J., Jeong, Y. J., Kwak, S. H. & Kim, T. S. Bilateral bronchiectasis and bronchiolitis at thin-section CT: diagnostic implications in nontuberculous mycobacterial pulmonary infection. Radiology 235, 282–288 (2005).
Song, J. W., Koh, W. J., Lee, K. S., Lee, J. Y., Chung, M. J., Kim, T. S. et al. High-resolution CT findings of Mycobacterium avium-intracellulare complex pulmonary disease: correlation with pulmonary function test results. AJR Am. J. Roentgenol. 191, 1070 (2008).
Guo, S. W. & Thompson, E. A. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48, 361–372 (1992).
Lin, S., Cutler, D. J., Zwick, M. E. & Chakravarti, A. Haplotype inference in random population samples. Am. J. Hum. Genet. 71, 1129–1137 (2002).
Watson, M. S., Cutting, G. R., Desnick, R. J., Driscoll, D. A., Klinger, K., Mennuti, M. et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet. Med. 6, 387–391 (2004).
Lee, J. H., Choi, J. H., Namkung, W., Hanrahan, J. W., Chang, J., Song, S. Y. et al. A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases. Hum. Mol. Genet. 12, 2321–2332 (2003).
Ngiam, N. S., Chong, S. S., Shek, L. P., Goh, D. L., Ong, K. C., Chng, S. Y. et al. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in Asians with chronic pulmonary disease: a pilot study. J. Cyst. Fibros. 5, 159–164 (2006).
Anzai, C., Morokawa, N., Okada, H., Kamidono, S., Eto, Y. & Yoshimura, K. CFTR gene mutations in Japanese individuals with congenital bilateral absence of the vas deferens. J. Cyst. Fibros. 2, 14–18 (2003).
Lee, K. H., Ryu, J. K., Yoon, W. J., Lee, J. K., Kim, Y. T. & Yoon, Y. B. Mutation analysis of SPINK1 and CFTR gene in Korean patients with alcoholic chronic pancreatitis. Dig. Dis. Sci. 50, 1852–1856 (2005).
Cuppens, H., Lin, W., Jaspers, M., Costes, B., Teng, H., Vankeerberghen, A. et al. Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes. The polymorphic (Tg)m locus explains the partial penetrance of the T5 polymorphism as a disease mutation. J. Clin. Invest. 101, 487–496 (1998).
Ghanem, N., Costes, B., Girodon, E., Martin, J., Fanen, P. & Goossens, M. Identification of eight mutations and three sequence variations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 21, 434–436 (1994).
Acknowledgements
This work was supported by Mid-career Researcher Program through a NRF grant funded by MEST (2011-0015546) and a Samsung Biomedical Research Institute grant (No. SBRI C-B1-101).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Jang, MA., Kim, SY., Jeong, BH. et al. Association of CFTR gene variants with nontuberculous mycobacterial lung disease in a Korean population with a low prevalence of cystic fibrosis. J Hum Genet 58, 298–303 (2013). https://doi.org/10.1038/jhg.2013.19
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.19
