
Abstract
Beta-thalassemia is one of the most prevalent inherited diseases and a public health problem in Malaysia. Malaysia is geographically divided into West and East Malaysia. In Sabah, a state in East Malaysia, there are over 1000 estimated cases of β-thalassemia major patients. Accurate population frequency data of the molecular basis of β-thalassemia major are needed for planning its control in the high-risk population of Sabah. Characterization of β-globin gene defects was done in 252 transfusion dependent β-thalassemia patients incorporating few PCR techniques. The study demonstrates that β-thalassemia mutations inherited are ethnically dependent. It is important to note that 86.9% of transfusion-dependent β-thalassemia major patients in Sabah were of the indigenous population and homozygous for a single mutation. The Filipino β0-deletion was a unique mutation found in the indigenous population of Sabah. Mutations common in West Malaysia were found in 11 (4.3%) patients. Four rare mutations (Hb Monroe, CD 8/9, CD 123/124/125 and IVS I-2) were also found. This study is informative on the population genetics of β-thalassemia major in Sabah.
Similar content being viewed by others

Introduction
Geographically, Malaysia is divided into West Malaysia (Peninsular Malaysia) and East Malaysia. The population is over 28.3 million and multiethnic,1 where ∼80% are from West Malaysia and 20% are from East Malaysia (Sabah and Sarawak).2 In East Malaysia, the population of Sabah and Sarawak are 70 and 50% indigenous, respectively. The population in Sabah was 3 117 405 in the year 2013,3 and divided into 35 officially recognized ethnic groups.4 The racial groups are heterogeneous where the largest indigenous ethnic group in Sabah is the Kadazandusun (25%), followed by Bajau (15%) and Murut (3%).3
Beta-thalassemia major (β-TM) is the most common inherited disorder of hemoglobin synthesis and a recognized public health problem in Malaysia.5, 6 The total number of transfusion-dependent thalassemia patients was 4768 in May 2010 (National Thalassaemia Registry), where the estimated transfusion dependent β-TM in Sabah was over 1000 cases.
β-thalassemia is characterized by a quantitative deficiency of functional β-globin chains, leading to an imbalanced globin chain production and an excess of α-globin chains. Precipitation of excess α-globin chains results in damage of the red cell precursor membrane leading to apoptosis and extensive intramedullary destruction of erythroid precursors in the bone marrow (ineffective erythropoiesis).7, 8 Wide spectrum of disease severity is a consequence of affected genes by different allele/mutations or different combinations of mutations inherited.9 β-thalasssaemia allele is the most consistent genotypic factor in prediction of phenotype of thalassemia.10 There are over 200 mutations reported that affect transcription, translation or RNA processing. Carriers or heterozygous for β-globin gene mutations are clinically normal and go through their life unaware of their carrier status. However, they have a 25% risk of having a child with β-TM in each pregnancy when both partners are β-thalassemia carriers. β-TM patient is transfusion dependent and requires iron chelation for life.11, 12 Mortality and morbidity in β-TM patients can be caused by the complications of iron overload, including sequelae of anemia, ineffective erythropoiesis and chelation therapy.12, 13
Each ethnic group has a common set of four to five mutations that comprises more than 95% of the mutations seen.11 In the last 15 years, the β-globin mutations has been well documented in the main racial groups in Peninsular Malaysia (West Malaysia) (Malays and Chinese5, 6, 14, 15, 16, 17, 18, 19, 20). The common β-globin mutations that account for 73% of the mutations in Malays are Hb E, IVS I-5 (G→C) and IVS I-1 (G→T). The common β-globin mutations that account for 90% of the mutations in Chinese-Malaysians are CD 41/42 (-TCTT), IVS II-654 (C→T), -28 (A→G), CD 17 (A→T) and CD 71/72 (+A), respectively.5, 6, 14, 15, 16, 17, 18, 19, 20 The complete spectrum of β-thalassemia mutations in East Malaysia is still unknown. Multiethnic migration from Brunei, Philippines and other places to this region makes the identification of β-thalassemia more complex. However, previous studies in limited number of β-TM patients from Sabah indicated the large β-globin gene deletion (Filipino β°-deletion) is the most common mutation found.20, 21, 22
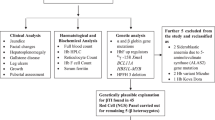
In this report, mutations in transfusion dependent β-TM patients from Sabah population were delineated using molecular analysis. As Filipino β0-deletion was a common mutation noted in previous studies, the patient’s DNA was first analyzed by Gap-PCR, followed by reverse dot blot hybridization and amplification refractory mutation system for mutations frequently encountered in West Malaysia. DNA sequencing was done to identify unknown mutations. In developing countries including Malaysia, technology for molecular analysis of thalassemia is limited. Elucidation of the spectrum of mutations that are largely ethnic-specific in their distribution will be informative for development of cost-effective and feasible PCR-based methods to identify β-thalassemia in this region.
Materials and methods
Study subjects
In this study, a total of 252 blood samples were collected from transfusion-dependent β-TM patients from 11 hospitals in different regions of Sabah. Approval was obtained from Medical Ethics Committee of the Faculty of Medicine and Health Sciences, Universiti Putra Malaysia (UPM/FPSK/PADS/T7-MJKEtikaPer/F01(JPAT_JUL(10)06)) and the Ministry of Health Medical Research Ethics Committee (NMRR-10-850-7075). The study was conducted in concordance with the declaration of Helsinki. Written informed consent was obtained from the study participants before blood sample collection.
DNA isolation
Two milliliters of venous blood was collected in ethylenediamine tetraacetic acid (EDTA) vacutainers from β-TM patients. Genomic DNA was extracted from leukocytes in peripheral whole blood samples by using QIAamp DNA midi kit (Qiagen GmbH, Hilden, Germany). Quality and quantity of the extracted genomic DNA were determined using Nanodrop 1000 Spectrophotometer (Thermo Scientific, Thermo Fisher Scientific Inc., Wilmington, DE, USA) and gel electrophoresis using 0.8% ethidium bromide-stained agarose gel in 1 × tris-acetic-EDTA buffer at 10 V cm−1 for 20 min.
Beta globin gene analysis
Filipino β0-deletion: Gap-PCR
Gap-PCR technique was performed by using specific primers that flank across the deleted region of the β-globin gene complex according to previous published assay.23 PCR amplification was conducted in 25 μl reaction volume containing 100 ng genomic DNA, a mixture of 2.5 mM MgCl2, 0.4 μM of the two forward primers, 0.6 μM of reverse primer, 50 mM KCl, 10 mM Tris (pH 8.3), 200 μM of mixture dNTP, 0.02 U μl−1 Taq polymerase and 2% glycerol. The cycling reaction was performed in a programmable thermal cycler (Takara PCR thermal cycler Dice, TP600 gradient, Takara Bio Inc., Otsu, Shiga, Japan) at initial denaturation for 5 min at 95 °C, followed by 35 cycles of 95 °C denaturation for 1 min, 60 °C annealing for 1 min, 72 °C extension for 1 min and a final extension for 10 min at 72 °C. Each amplified product (10 μl) was analyzed using 1.5% ethidium bromide-stained agarose gel in 1X TAE buffer at 10 volts cm−1 for an hour. The gel was then visualized on an ultraviolet transilluminator (G:box bioimaging systems, Synoptics Ltd, Cambridge, UK). If Gap-PCR failed to detect the mutation, the samples were further analyzed for point mutations.
Point mutations detection: reverse dot blot hybridization-strip assay
A total of 33 samples consisting of 22 β-TM heterozygous for the Filipino β0-deletion and 11 with unidentified mutations were further analyzed for another 22 β-thalassemia mutations using β-globin strip assay (Viennalab Diagnostics GmbH, Vienna, Austria). The 22 β-globin mutations screened for were: -31 (A→G) [NM_000518.4:c.-50-31A>G]; -29 (A→G) [NM_000518.4:c.-50-29A>G]; -28 (A→G) [NM_000518.4:c.-50-28A>G]; CAP+1 (A→C) [NM_000518.4:c.-50A>C]; Init CD ATG>AGG [NM_000518.4:c.2T>G]; CD 8/9 (+G) [NM_000518.4:c.27_28insG], CD 15 (TGG→TAG) [NM_000518.4:c.47G>A]; CD 17 (A→T) [NM_000518.4:c.52A>T];CD 19 (AAC→AGC; Asn→Ser; Hb Malay) [NM_000518.4:c.59A>G]; CD 26 (G→A; Glu→Lys; Hb E) [NM_000518.4:c.79G>A]; CD 27/28 (+C) [NM_000518.4:c.84_85insC]; IVS I-I (G→T) [NM_000518.4:c.92+1G>T]; IVS I-5 (G→C) [NM_000518.4:c.92+5G>C]; CD 41/42 (-TTCT) [NM_000518.4:c.124_127delTTCT]; CD 43 (GAG→TAG) [NM_000518.4:c.130G>T]; CD 71/72 (+A) [NM_000518.4:c.216_217insA]; CD 89/9 (-GT)[NM_000518.4:c.269_270delGT]; CD 90 (GAG→TAG) [NM_000518.4:c.271G>T]; CD 95 (+A) [NM_000518.4:c.287_288insA]; IVS II-1 (G→A) [NM_000518.4:c.315+1G>A]; IVS II-654 (C→T) [NM_000518.4:c.316-197C>T] and CD 121 (GAA→TAA; Glu→TermCD) [NM_000518.4:c.364G>T].
Point mutations detection: amplification refractory mutation system-PCR
amplification refractory mutation system-PCR was run in parallel for the detection of the 12 common β-thalassemia mutations seen in Malaysia24, 25 (Hb E, Hb Malay, IVS I-5 (G→C), IVS I-1 (G→T), CD 8/9 (+G), CAP+1 (A→C), CD 41/42 (-TTCT), IVS II-654 (C→T), CD 17 (A→T), -28 (A→G), -29 (A→G) and CD 71/72 (+A)).
When the reverse dot blot hybridization and amplification refractory mutation system-PCR assays were unable to detect any mutations, sequencing of β-globin gene was performed.
Unknown mutations detection: DNA Sequencing
The 1.6 kb of β-globin gene including the 5′ and 3′ untranslated region was amplified in three fragments. 5′ promoter region, 5′ untranslated region, exon 1, intron 1 and part of exon 2 were amplified using primer βA-Fw (5′-CGATCTTCAATATGCTTACCAA-3′) and βA-Rv (5′-AACGATCCTGAGACTTCCACA-3′) as a 947 bp fragment. Intron 2 was amplified using primer βB-Fw (5′-GCACGTGGATCCTGAGAACT-3′) and βB-Rv (5′-CACACAGACCAGCACGTTG-3′) as a 901 bp fragment. Exon 3 and 3′ untranslated region were amplified using primer βC-Fw (5′-GCTAATCATGTTCATACCTCTT-3′) and βC-Rv (5′-CAGATTCCGGGTCACTGTG-3′) as a 854 bp fragment.26, 27 PCR amplification was conducted in 50 μl reaction volume containing 100 ng genomic DNA, 2.5 mM MgCl2, 0.4 μmol of each primer, 50 mM KCl, 10 mM tris (pH 8.3), 200 μM of each dNTP and 0.04 U μl−1 Taq polymerase. The cycling reaction was performed in a programmable thermal cycler (Takara PCR thermal cycler Dice, TP600 gradient, Takara Bio Inc.) at initial denaturation for 5 min at 95 °C, followed by 35 cycles of 95 °C denaturation for 30 s, 60 °C annealing for 30 s, 72 °C extension for 1 min and a final extension for 7 min at 72 °C. PCR products were purified using the QIAquick PCR purification kit (Qiagen), followed by quality and quantity analysis using Nanodrop 1000 Spectrophotometer. Pure PCR products were sent for sequencing service using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Warrington, UK) and analyzed on the ABI 3100 Genetic Analyzer (Applied Biosystems). Sequence alignment and assembly was done by using DNA baser sequence assembly software (Heracle Biosoft SRL, Arges, Germany).
Results
A total of 252 β-TM patients from Sabah were recruited for this study. Frequency of β-thalassemia alleles in β-TM patients in Sabah population is shown in Table 1, whereas the ethnic distribution among the β-TM patients is shown in Table 2. Filipino β0-deletion was identified as the most common mutation with 460 alleles (91.3%). A total of 219 (86.9%) patients were homozygous for the Filipino β0-deletion: 185 (84.5%) were from the indigenous population; Kadazandusun (137), Rungus (20), Murut (10), Sungai (9), Bajau (4) and Bruneian (5). Two were found with mixed-indigenous ethnicity (indigenous-Sabah/Chinese (2)). The ethnicities for the remaining 32 β-TM patients homozygous for Filipino β0-deletion were not reported.
Compound heterozygosity of Filipino β0-deletion was found in 22 (8.7%) patients where 16 patients (72.7%) had both Filipino β0-deletion and β+-mutations (β°/β+). Among the 22 cases, 6 were found compound heterozygous with CD 26 (G→A; Hb E) and CD 19 (A→G; Hb Malay), respectively. Two were compound heterozygous with poly-A tail mutation (AATAAA→AATAGA) and CD 71/72 (+A), respectively. The remaining four cases were compound heterozygous for CD 17 (A→T), IVS I-5 (G→C), IVS II-654 (C→T) and one rare mutation, Hb Monroe (G→C). The second mutation for two cases could not be identified. Mutations common in West Malaysia were found in 11 (4.3%) patients.
Discussion
Sabah, a state in Malaysia, is situated at the northern part of the Borneo Island. In this report, we describe the spectrum of β-globin gene mutations identified in 252 β-TM patients in Sabah. The mutations identified in this region showed great discrepancy when compared with the West Malaysia (Peninsular Malaysia).5, 6, 14, 15, 16, 17, 18, 19, 20 These findings indicate a clear genetic difference between Sabah (East Malaysia) and West Malaysia. This can be attributed by regional differences in demography and that the population in Sabah and Peninsular Malaysia is genetically distinct historically.
The close proximity of Sabah in North Borneo and the Philippines has allowed migration drift to occur. The Filipino β0-deletion was first reported by Motum et al.28 in a Filipino family and found in 45.8% of the β-globin mutant alleles in the Taiwan-Filipinos.28, 29 This mutation was also found in Indonesians from the eastern part of Indonesia due to its adjacent geographical locations to Philippines.30, 31 In our study, Filipino β0-deletion was the predominant mutation identified in the indigenous population of Sabah (homozygosity: 219 (86.9%) especially in Kadazandusun (137 (62.6%)). This is in keeping with previous studies, where the Fililpino β0-deletion was also found in the Kadazandusun.21, 22, 32 We conclude that Filipino β0-deletion is largely specific in its distribution, occurring predominantly in the indigenous population of Sabah.
In β-thalassemia, the majority of mutations are point mutations (such as single nucleotide substitutions), minor insertions or deletions within the gene or its immediate flanking sequence.7, 8 The Fililpino β0-deletion is the largest deletion seen among the mutations identified in Malaysia, which leads to a severe β0-thalassemia phenotype with totally absence of β-globin chains expression. This deletion was first reported as 45 kb deletion17, 23 and in a recent study, it was discovered as 118 kb deletion.33 The 5′ deletion breakpoint had been defined at the position −4279 relative to the mRNA capsite of the β-globin gene and the 3′ breakpoint extending to the downstream of β-globin gene, six olfactory reception genes (four functional OR genes and two OR pseudogenes) including one γ-globin enhancer located at OR52A1. This γ-globin enhancer contained enhancer element that conveys continuous expression of fetal globin genes. Deletion of this enhancer might contribute to a significant reduction of the compensatory γ-globin gene expression, leading to lower Hb F production and exacerbate the phenotype of β-thalassemia.23, 28, 33
Common hemoglobin (Hb) variants in West Malaysia were found to coexist with the Fililpino β0-deletion. The Hb variants found in our study were Hb Malay and Hb E. Hb E is a common β+-Hb variant found among Malays.6 Coexistence of Hb E with Filipino β0-deletion had been reported in Thai population34 and three Indonesian patients.31 The Thai patient had severe hypochromic microcytic anemia and an elevation of Hb A2 level. In our study six patients were found to coexist with Hb E, where only one was a Kadazandusun. The presence of Hb E mutation in Sabah may be due to intermarriage of Malays with the indigenous population.
Coexistence of Filipino β0-deletion with some common mutations in West Malaysia was also found. Common β-thalassemia mutations were seen in seven (2.8%) of β-TM patients, consisting of Poly-A (A→G); CD 71/72 (+A); CD 17 (A→T); IVS I-5 (G→C) and IVS II-654 (C→T). We may assume that the Filipino β0-deletion allele was inherited from the parent who is from the indigenous population, whereas the second allele might be inherited from the parent who is Malaysian-Chinese or Malaysian-Malay from West Malaysia. However, further studies into ethnicity background in this group would be required.
In the transfusion dependent β-TM patients, 16 (72.7%) were β°/β+. These patients may have been clinically severe as a consequence of gene modifiers. Environmental factors, intercurrent infections, nutritional status and access to health care facilities may be confounding factors.35
Eleven patients (4.4%) were found without the Filipino β0-deletion allele and no Kadazandusun were found in this group. The majority were Chinese, followed by Malay, Bruneian, Bajau and Banjar. Among the six Chinese patients, the mutations inherited were in concordance with the common mutations found among Malaysian-Chinese in West Malaysia.5, 6, 14, 15, 16, 17, 18, 19, 20 Similar observation was noticed in the Malay patient with Hb E/IVS I-5, which are common mutations among Malaysian-Malays in West Malaysia.5, 6, 14, 15, 16, 17, 18, 19, 20 The remaining four patients inherited some rare mutations (Hb Monroe, CD 123/124/125, IVS I-2 (T→G) and CD 8/9 (+G)), which were rarely found in West Malaysia.
The patient who was compound heterozygous for Filipino β0-deletion with Hb Monroe originated from Jawa. This rare mutation was first reported in 1988 by Gonzalez-Redondo et al. in a transfusion-dependent 15-year-old black female from USA and is commonly found in Black, Libyan, Tunisian, Mediterranean and African-Americans.36, 37, 38 Hb Monroe (NM_000518.4:c.92G>C) also known as Hb Kairouan and has a β0-thalassemia phenotype. The mutation is located at codon 30 of exon 1 with position at chromosome 11, 5 248 160. In the normal β-globin gene, codon 30 (AGG) is divided by intron 1 and AG dinucleotide is required for normal splicing. The change of nucleotide G to C at position −1 of intron 1 results in transition of Arg to Thr (AGG→ACG), forming the mutant hemoglobin (Hb Monroe), which severely reduces the utilization of the normal 5′ splice site. The splicing efficiency will be reduced as 98% of the correct splicing is inhibited and no normal mRNA is formed (Itha ID: 100; Hb Var ID: 290).37, 38, 39, 40
Compound heterozygous of CD 123/124/125 with CD 19 was found in an indigenous patient with Banjar ethnicity. This mutation was first reported in 1991 by Fucharoen et al.41 in a 3-year-old Northeastern Thai patient with large inclusion bodies observed in peripheral blood. This variant is a β0-thalassemia with deletion of eight bases (5′-ACCCCACC-3′) in CD 123/124/125, exon 3 of β-globin gene situated at chromosome 11, from 5 246 894–5 246 902 (NM_000518.4:c.370_378delACCCCACCA). This mutation is also known as beta-Khon Kaen and results in formation of big inclusion bodies due to the changes of the reading frame of 135 amino acids with the new codon 136, which is a stop codon (TAA). The normal amino-acid residues of the β-globin chain are eliminated from codon 123–146, where the H-helix involved in α1β1 contact and α1β2 subunit interactions will be interfered. The mutant globin chain does not interact with the α-globin chain and will be removed by proteolysis. Hence, the chains produced are highly unstable and are likely to be degraded soon after translation (Itha ID: 246; Hb Var ID: 953).39, 40
Compound heterozygous of Hb E with IVS I-2 (T→G; AGGTTGGT→AGGGTGGT) (NM_000518.4:c.92+2T>G) was found in an indigenous patient with Bajau ethnicity. IVS I-2 is commonly found in Tunisians. This rare mutation results in β0-thalassemia with the transition of T to G at intron 1 with position at chromosome 11, 5 248 158. The transition changes in the GT dinucleotide disturbs the normal splicing event and no normal mRNA is produced (Itha ID: 104; Hb Var ID: 821).39, 40
In our study, compound heterozygous CD 8/9 (+G) with poly-A (A→G) were seen in two Bruneian patients. This mutation is commonly found in Asian Indians, such as Pathans with the frequency of 48.3%,42 followed by Pakistani (25.9%), Punjabi (31.9%42), Iranian (11.03%), Bangladeshi (10%) and Spanish (8.6%43). CD 8/9 (+G) (NM_000518.4:c.27_28insG) results by insertion of a G nucleotide between the codons 8 and 9 of β-gene, located at exon 1 of chromosome 11, at 5 248 224–5 248 225. It changes the sequences AAG TGT (Lys; Ser) to AAG G TCT. This frameshift results in the termination of translation at codon 22 (TGA) and has a β0-thalassemia phenotype with complete absence of β-globin chain production (Itha ID: 62; Hb Var ID: 786).39, 40, 42, 43, 44
Conclusion
Genotype characterization in thalassemia is crucial to the successful implementation of prenatal diagnosis and disease management strategies. Ethnicity is an important factor in the molecular diagnosis of β-thalassemia. In this study, the spectrum of β-thalassemia mutations in Sabah was delineated. This report reveals a notable regional specificity of the Filipino β0-deletion. Given the demonstrated low frequency of β-thalassemia alleles in the indigenous population of Sabah, the present study is informative. This finding suggests that the indigenous population of Sabah and Philippines may belong to the same stock and origin. Importantly testing for the Filipino β0-deletion as the first step identified this mutation in 86.9% of β-TM patients in Sabah. Thus, a cost-effective strategy would be to do Gap-PCR for the Filipino β0-deletion as the first step to identify mutations in β-TM patients.
References
Population distribution and basic demographic characteristic report Department of Statistics Malaysia [homepage on the internet]. c2010 [updated 2013 Sept 11; cited 2013 Sept 12]. Available from http://www.statistics.gov.my/portal/index.php?option=com_content&id=1215&Itemid=89&lang=en (2010).
ICID—Irrigation & Drainage in the World—A Global Review. pp 1-6 [homepage on the Internet]. c2010 [updated 2013 Sept 11; cited 2013 Sept 12]. Available from http://www.icid.org/i_d_malaysia.pdf.
Total population by ethnic group, administrative district and state, Malaysia Department of Statistics Malaysia [homepage on the Internet]. c2010 [updated 2013 Sept 11; cited 2013 Sept 12]. Available from http://www.statistics.gov.my/portal/download_Population/files/population/05Jadual_Mukim_negeri/Mukim_Sabah.pdf (2010).
Borneo Hidden Treasures website [homepage on the Internet]. c2013 [updated 2010 May; cited 2013 Sept 12]. Available from http://goeasytravel.co/get/?page_id=741.
Tan, J. A. M. A., George, E., Tan, K. L., Chow, T., Tan, P. C., Hassan, J. et al. Molecular defects in the β-globin gene identified in different ethnic groups/populations during prenatal diagnosis for β-thalassaemia: a Malaysian experience. Clin. Exp. Med. 4, 142–147 (2004).
George, E., Li, H. J., Fei, Y. J, Reese, A. L., Baysal, E., Cepreganova, B. et al. Types of thalassameia among patients attending a large university clinic in Kuala Lumpur, Malaysia. Hemoglobin 16, 51–66 (1992).
Thein, S. L. Genetic insights into the clinical diversity of β thalassaemia. Br. J. Haematol. 124, 264–274 (2004).
Thein, S. L. Pathophysiology of β thalassemia-a guide to molecular therapies. Hematology Am. Soc. Hematol. Edu. Program 1, 31–37 (2005).
Bowden, D. K. Abnormal laboratory results: screening for thalassaemia. Aust. Prescr. 24, 120–123 (2001).
Ho, P., Hall, G., Luo, L., Weatherall, D. & Thein, S. Beta-thalassaemia intermedia: is it possible consistently to predict phenotype from genotype? Br. J. Haematol. 100, 70–78 (1998).
George, E. Beta thalassaemia major in Malaysia, an on-going public health problem. Med. J. Malaysia 56, 397–400 (2001).
Quek, L. & Thein, S. L. Molecular therapies in β–thalassaemia. Br. J. Haematol. 136, 353–365 (2006).
Fucharoen, S. & Winichagoon, P. Prevention and control of thalassemia in Asia. Asian Biomed. 1, 1–6 (2010).
Sivalingam, M., Looi, M., Zakaria, S., Hussin, N., Alias, H., Latiff, Z. et al. Molecular study and genotype/phenotype correlation of β thalassemia in Malaysia. Int. J. Lab. Hematol. 34, 1–6 (2012).
George, E., George, R., Ariffin, W. A., Mokhtar, A. B., Azman, Z. A. & Sivagengei, K. Spectrum of beta-thalassaemia mutations in transfusion dependent thalassaemia patients: practical implications in prenatal diagnosis. Med. J. Malaysia 48, 325–329 (1993).
George-Kodiseri, E., Yang, K. G., Kutlar, F., Wilson, J. B., Kutlar, A., Stoming, T. A. et al. Chinese in west Malaysia: the geography of beta thalassaemia mutations. Singapore Med. J. 31, 374–377 (1990).
Yang, K. G., Kutlar, F., George, E., Wilson, J. B., Kutlar, A., Stoming, T. A. et al. Molecular characterization of β–globin gene mutations in Malay patients with Hb E–β–thalassaemia and thalassaemia major. Br. J. Haematol. 72, 73–80 (1989).
George, E., Huisman, T. H., Yang, K. G., Kutlar, F., Wilson, J. B., Kutlar, A. et al. First observation of haemoglobin Malay alpha 2B2 26 (B1) Asn—Ser—a case report. Med. J. Malaysia 44, 259–262 (1989).
George, E. The clinical severity of beta-thalassemia mutations in west Malaysia. Southeast Asian J. Trop. Med. Public Health 26, 225 (1995).
Tan, J. A. M. A., Chin, P. S., Wong, Y. C., Tan, K. L., Chan, L. L. & George, E. Characterisation and confirmation of rare beta-thalassaemia mutations in the Malay, Chinese and Indian ethnic groups in Malaysia. Pathology 38, 437–441 (2006).
Thong, M. K. & Soo, T. The spectrum of beta-globin gene mutations in children with beta-thalassaemia major from Kota Kinabalu, Sabah, Malaysia. Singapore Med. J. 46, 340–343 (2005).
Thong, M. K., Rudzki, Z., Hall, J., Tan, J. A., Chan, L. L. & Yap, S. F. A single, large deletion accounts for all the beta-globin gene mutations in twenty families from Sabah (north Borneo), Malaysia. Hum. Mutat. 13, 413 (1999).
Waye, J. S., Eng, B., Hunt, J. A. & Chui, D. H. K. Filipino β-thalassemia due to a large deletion: identification of the deletion end points and polymerase chain reaction (PCR)-based diagnosis. Hum. Genet. 94, 530–532 (1994).
Old, J. M. Detection of mutations by the amplification mutation system Prot. Human Mol. Genet. Methods Mol. Bio. 9, 77–84 (1991).
Old, J. M. Screening and genetic diagnosis of haemoglobin disorders. Blood Rev. 17, 43–53 (2003).
Clark, B. & Thein, S. Molecular diagnosis of haemoglobin disorders. Clin. Lab. Haematol. 26, 159–176 (2004).
Knott, M., Ramadan, K., Savage, G., Jones, F., El-Agnaf, M., McMullin, M. et al. Novel and mediterranean β thalassemia mutations in the indigenous northern Ireland population. Blood Cells Mol. Dis. 36, 265–268 (2006).
Motum, P, Kearney, A, Hamilton, T & Trent, R. Filipino beta zero thalassaemia: a high Hb A2 beta zero thalassaemia resulting from a large deletion of the 5'beta globin gene region. J. Med. Genet. 30, 240–244 (1993).
Ko, T. M., Caviles, A. Jr, Hwa, H. L., Liu, C. W., Hsu, P. M. & Chung, Y. P. Prevalence and molecular characterization of β-thalassemia in filipinos. Ann. Hematol. 77, 257–260 (1998).
Setianingsih, I., Williamson, R., Marzuk, S., Harahap, A., Tamam, M. & Forrest, S. Molecular basis of β-thalassemia in Indonesia: application to prenatal diagnosis. Mol. Diagn. 3, 11–20 (1998).
Setianingsih, I., Williamson, R., Daud, D., Harahap, A., Marzuki, S. & Forrest, S. Phenotypic variability of filipino β°-thalassemia/HbE patients in Indonesia. Am. J. Hematol. 62, 7–12 (1999).
Tan, J. A. M. A., Lee, P. C., Wee, Y. C., Tan, K. L., Mahali, N. F., George, E. et al. High prevalence of alpha-and beta-thalassemia in the kadazandusuns in east Malaysia: challenges in providing effective health care for an indigenous group. J. Biomed. Biotechnol. 706872, 1–5 (2010).
Van Ziffle, J., Yang, W. & Chehab, F. F. Homozygous deletion of six olfactory receptor genes in a subset of individuals with beta-thalassemia. PLoS One 6, e17327 (2011).
Yamsri, S., Sanchaisuriya, K., Fucharoen, G. & Fucharoen, S. Genetic origin and interaction of the filipino β0-thalassemia with Hb E and α-thalassemia in a Thai family. Transl. Res. 159, 473–476 (2012).
George, E. HbE β-thalassaemia in Malaysia: revisited. J. Hematol. Thromb. Dis. 1, 2 (2013).
Gonzalez-Redondo, J., Stoming, T., Lanclos, K., Gu, Y., Kutlar, A., Kutlar, F. et al. Clinical and genetic heterogeneity in black patients with homozygous beta-thalassemia from the southeastern united states. Blood 72, 1007–1014 (1988).
Moosa, M. M., Ayub, M. I., Bashar, A. M. A. E., Sarwardi, G., Khan, W., Khan, H. et al. Combination of two rare mutations causes β-thalassaemia in a bangladeshi patient. Genet. Mol. Biol. 34, 406–409 (2011).
Vidaud, M., Gattoni, R., Stevenin, J., Vidaud, D., Amselem, S., Chibani, J. et al. A 5'splice-region G→C mutation in exon 1 of the human beta-globin gene inhibits pre-mRNA splicing: A mechanism for β+-thalassemia. Proc. Natl Acad. Sci. 86, 1041–1045 (1989).
Haemoglobin Variants Database of human haemoglobin Variants and thalassaemia mutations [homepage on the internet]. c2013 [updated 2013 Sept; cited 2013 Sept 12]. Available from http://globin.bx.psu.edu/hbvar.
Ithanet Database of Ithnanet Portal [homepage on the internet]. c2013 [updated 2013 Sept; cited 2013 Sept 12]. Available from http://www.ithanet.eu/db/ithagenes.
Fucharoen, G., Fuchareon, S., Jetsrisuparb, A. & Fukumaki, Y. Eight-base deletion in exon 3 of the beta-globin gene produced a novel variant (beta khon kaen) with an inclusion body beta-thalassemia trait. Blood 78, 537–539 (1991).
Khattak, S. A. K., Ahmed, S., Anwar, J., Ali, N. & Shaikh, K. H. Prevalence of various mutations in beta thalassaemia and its association with haematological parameters. J. Pak. Med. Assoc. 62, 40–43 (2012).
Villegas, A., Ropero, P., Gonzalez, F. A., Martí, E., Anguita, E. & De Blas, J. High incidence of the CD8/9 (+G) beta0-thalassemia mutation in Spain. Haematologica 83, 1066–1068 (1998).
George, E., Teh, L. K., Lai, M. I. & Tan, J. A. M. A. Beta thalassaemia mutations in Malays: a simplified cost-effective strategy to identify the mutations. MJMHS 8, 45–53 (2012).
Acknowledgements
We thank the Director General of Health, Malaysia for permission to publish this paper (NMRR-10-850-7075). This study was supported by E-Science Research Grant, Ministry of Science, Technology and Innovation (MOSTI) 02-01-04-SF1031 awarded to George Elizabeth.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Teh, L., George, E., Lai, M. et al. Molecular basis of transfusion dependent beta-thalassemia major patients in Sabah. J Hum Genet 59, 119–123 (2014). https://doi.org/10.1038/jhg.2013.131
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.131
Keywords
This article is cited by
-
Evidence of new intragenic HBB haplotypes model for the prediction of beta-thalassemia in the Malaysian population
Scientific Reports (2021)
-
A commentary on molecular basis of transfusion dependent beta-thalassemia major patients in Sabah
Journal of Human Genetics (2014)
