
Abstract
Yunis–Varon syndrome (YVS, MIM 216340) is a rare autosomal recessive disorder characterized by skeletal abnormalities and severe neurological impairment with vacuolation of the central nervous system, skeletal muscles and cartilages. Very recently, mutations of the FIG4 (FIG4 homolog, SAC1 lipid phosphatase domain containing (Saccharomyces cerevisiae)) gene, which encodes a 5′-phosphoinositide phosphatase essential for endosome/lysosome function have been identified as the cause for YVS. Interestingly, FIG4 mutations were previously reported to be responsible for other neurodegenerative diseases such as autosomal recessive Charcot–Marie–Tooth disease type 4J and autosomal dominant amyotrophic lateral sclerosis/primary lateral sclerosis. We analyzed a YVS patient using whole-exome sequencing, and identified novel biallelic FIG4 mutations: c.1750+1delG and c.2284_2285delCT (p.S762Wfs*3). These two mutations were mutations supposed to have null function. To our knowledge, this is the second report of FIG4 mutations in YVS and our result supports the idea that biallelic null mutations of FIG4 cause YVS in human.
Similar content being viewed by others
Main
Yunis–Varon syndrome (YVS, MIM_216340) is a rare autosomal recessive disorder characterized by skeletal abnormalities (cleidocranial dysostosis, bilateral absence of thumbs and halluces distal aphalangia and pelvic bone dysplasia) and severe neurological impairment.1 Recently, mutations of FIG4 (FIG4 homolog, SAC1 lipid phosphatase domain containing (Saccharomyces cerevisiae); NM_014845.5) were identified as the genetic cause for YVS.2 FIG4 encodes FIG4 protein (also known as SAC3) which is a 5′-phosphoinositide phosphatase essential for endosome/lysosome function.3 FIG4 binds with Vac14/ArPIKfyve and Fab1/PIKfyve to form a functional complex on early endosomal membranes known as PIKfyve–ArPIKfyve–Sac3 complex.3 The PIKfyve–ArPIKfyve–Sac3 complex mediates the conversion of endosomal phosphatidylinositol 3-phosphate (PI3P) to phosphatidylinositol 3,5-biphosphate (PI(3,5)P2), and this conversion is essential for protein sorting, trafficking late endosomes to lysosomal degradation compartment and regulating some other endolysosome/lysosome functions essential for degradation such as ion channel activation and endolysosome fusion/fission.4 Impairment of FIG4 causes a reduction of PI(3,5)P2, which results in the malfunction of the endosome/endolysosome/lysosome. Therefore, the accumulation of undegraded materials in these compartments leads to dilatation.3
FIG4 abnormalities were previously reported to be the causative for autosomal recessive Charcot–Marie–Tooth disease type 4J (CMT4J, MIM#611228) and autosomal dominant amyotrophic lateral sclerosis (ALS, MIM#105400)/primary lateral sclerosis (PLS, MIM#611637).2, 5, 6, 7 Vacuolated endolysosomes are found in the perinuclear regions of peripheral neurons of Fig4-null mice and CMT4J patients.3 In YVS patients, vacuolation was observed in skeletal muscles, fibroblasts and the central nervous system including cerebral cortex, the basal ganglia, cerebellar olives and medullary olives.8, 9, 10
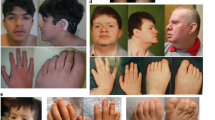
The patient was a 2-year-old girl at the time of genetic consultation. She was the second daughter of a nonconsanguineous pair of 32-year-old woman and a 25-year-old man. Both her parents and her sister were healthy. Fetal echogram revealed intrauterine growth retardation. She was born at 40 weeks of gestation, with Apgar scores of 2 and 1 at 1 and 5 min, respectively. Her birth weight was 3306 g (+0.6 SD), body length 52 cm (+1.5 SD) and head circumference 33 cm (−0.3 SD). Because of neonatal asphyxia, she was transferred to neonatal intensive care unit and supported by mechanical ventilation. Lower anal atresia was found and colostomy was performed. Tube feeding was necessary because of poor feeding activity. Tracheostomy was placed at 8 months of age because of severe respiratory impairment. She presented with characteristic face (protruding forehead, wide fontanels, temporal narrowing, hypertelorism, blepharophimosis, inverted epicanthus, flat nasal bridge, bilateral low set ears, short philtrum, high-arched palate, down-turned mouth, micrognathia and sparse and depigmented scalp hair), hypoplasia of the right clavicle and other long bones, aplasia of the left clavicle, absence of bilateral thumbs and halluces, hypoplasia of the distal phalanges, multiple appendicular bone fracture and proctatresia (Figures 1a and c). Congenital cataract and hearing loss were also noted. Head computed tomography showed severe hypoplasia of the cerebrum and enlargement of lateral, third and fourth ventricles (Figure 1d). At the age of 2 years, her height was 67.4 cm (−4.9 SD), body weight was 9.1 kg (−1.9 SD) and head was circumference 46.2 cm (−0.2 SD). Her developmental mile stones were severely retarded. She was bedridden and unable to support her head. She was therefore diagnosed with YVS.

Imaging studies of the proband. (a) Thoracoabdominal X-ray on the first day after birth (dorsal position). Hypoplasia of both clavicle and the fracture of left humerus were evident. (b) X-ray of left forearm and hand. Absence of thumbs and hypoplastic phalangeal bones were observed. (c) X-ray of right foot. Absence of halluces and hypoplastic phalangeal bones was noted. (d) Head CT. Extended hypoplasia of the cerebrum and enlargement of the third and the lateral ventricles were noted. The fourth ventricle was also slightly enlarged.
To identify the genetic cause of the proband, we performed whole-exome sequencing on the patient and her parents as FIG4 mutations were not described in YVS when we started the genetic analysis. Peripheral blood samples were collected from the trio (patient and both parents) after obtaining written informed consent. This study was approved by the institutional review board of Yokohama City University School of Medicine. Each individual’s DNA was captured with the SureSelect Human All Exon v4 Kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on a HiSeq2000 with 101 bp paired-end reads and 7 bp index reads (Illumina, San Diego, CA, USA). Image analysis and base calling were performed by sequence control software real-time analysis and CASAVA software v1.8 (Illumina). The reads were aligned to a human reference genome (hg19) with Novoalign 2.08.02 (http://www.novocraft.com/). After the removal of PCR duplication by Picard, the variants were called by Genome Analysis Toolkit 1.6-5 (GATK: http://www.broadinstitute.org/gatk/) and annotated by ANNOVAR (2012feb) (http://www.openbioinformatics.org/annovar/). Through this flow, common variants registered in common dbSNP137 (minor allele frequency ⩾0.01) (http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=335665349&c=chr6&g=snp137Common) were removed.
More than 93% of coding sequence was covered by at least 20 reads in each individual. Because this syndrome was suspected as an autosomal recessive disease because of the consanguinity and/or affected siblings in reported families,1, 11, 12 we extracted homozygous or compound heterozygous variants using whole-exome sequencing data. Through data processing synonymous variants, variants in segmental duplications, variants registered in dbSNP137 or our in-house database (exome data of 408 Japanese individuals) were all excluded from the candidates (Figure 2). As a result, only one compound heterozygous mutation in FIG4 remained: c.1750+1delG and c.2285_2286delCT (p.S762Wfs*3), which were also confirmed by Sanger sequencing. The c.1750+1delG at intron 15 and c.2283_2284delCT at exon 20 were inherited from her mother and father, respectively (Figure 3).

Priority scheme of whole-exome sequencing data. Numbers of variants surviving after each selection are shown.

FIG4 mutations in the patient. (a) Pedigree and mutations. (b) Schematic presentation of the FIG4 protein and mutations found in the patient. (c) Electropherograms corresponding to each mutation in the patient, her father and mother. WT: wild-type allele, Mut: mutant allele. The altered or deleted bases were marked by square.
Campeau et al.2 proposed a model in which the clinical difference among the above-mentioned three conditions regarding FIG4 abnormalities (that is, YVS, CMT4J and ALS/PLS) depends on how much residual FIG4 function remains based on the following evidence: (1) all previously reported CMT4J patients had compound heterozygous mutations but carried one null allele and one missense allele, whereas all YVS patients had biallelic null mutations.7, 13, 14, 15 (2) ALS/PLS patients had heterozygous null or partially loss-of-function mutations.5 (3) In functional assays, CMT4J patients had less FIG4 activity than ALS/PLS patients.5 Biallelic null mutations in our YVS patient support this model.
The clinical effects of heterozygous FIG4 mutations remain unclear. Theoretically, all parents (excluding the patient having a de novo truncation mutation) of YVS patients should carry a heterozygous null mutation. However, none of the parents of identified CMT4J patients or YVS patients, who should be heterozygous carriers, have been reported to show ALS/PLS. The current age of the father and mother in this study were 27 and 34 years old, respectively, and they did not show any ALS/PLS phenotype. Furthermore, the patient’s grandparents did not show any sign of ALS/PLS. This discrepancy could be presymptomatic status of the family members carrying a heterozygous mutation as the average onset age of ALS/PLS in patients with FIG4 mutation was reported to be 56±14 years (mean±SD).5 It would be strongly encouraged to observe their longitudinal clinical course of heterozygous carriers in human. The other possibility for this discrepancy would be unknown genetic modifier(s) leading to the incomplete penetrance. To clarify the genotype–phenotype correlation and the diverse clinical phenotypes by FIG4 mutations, further studies of genetic and clinical features are necessary.
References
Yunis, E. & Varon, H. Cleidocranial dysostosis, severe micrognathism, bilateral absence of thumbs and first metatarsal bone, and distal aphalangia: a new genetic syndrome. Am. J. Dis. Child. 134, 649–653 (1980).
Campeau, P. M., Lenk, G. M., Lu, J. T., Bae, Y., Burrage, L., Turnpenny, P. et al. Yunis–Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am. J. Hum. Genet. 92, 781–791 (2013).
Martyn, C. & Li, J. Fig4 deficiency: a newly emerged lysosomal storage disorder? Prog. Neurobiol. 101-102, 35–45 (2013).
Huotari, J. & Helenius, A. Endosome maturation. EMBO J. 30, 3481–3500 (2011).
Chow, C. Y., Landers, J. E., Bergren, S. K., Sapp, P. C., Grant, A. E., Jones, J. M. et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 84, 85–88 (2009).
Zhang, X., Chow, C. Y., Sahenk, Z., Shy, M. E., Meisler, M. H. & Li, J. Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain 131, 1990–2001 (2008).
Nicholson, G., Lenk, G. M., Reddel, S. W., Grant, A. E., Towne, C. F., Ferguson, C. J. et al. Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P(2) phosphatase FIG4. Brain 134, 1959–1971 (2011).
Garrett, C., Berry, A. C., Simpson, R. H. & Hall, C. M. Yunis–Varon syndrome with severe osteodysplasty. J. Med. Genet. 27, 114–121 (1990).
Walch, E., Schmidt, M., Brenner, R. E., Emons, D., Dame, C., Pontz, B. et al. Yunis–Varon syndrome: evidence for a lysosomal storage disease. Am. J. Med. Genet. 95, 157–160 (2000).
Dworzak, F., Mora, M., Borroni, C., Cornelio, F., Blasevich, F., Cappellini, A. et al. Generalized lysosomal storage in Yunis Varon syndrome. Neuromuscul. Disord. 5, 423–428 (1995).
Rabe, H., Brune, T., Rossi, R., Steinhorst, V., Jorch, G., Horst, J. et al. Yunis–Varon syndrome: the first case of German origin. Clin. Dysmorphol. 5, 217–222 (1996).
Pfeiffer, R. A., Diekmann, L. & Stock, H. J. Aplasia of the thumbs and great toes as the outstanding feature of Yunis and Varon syndrome. A new entity. A new observation. Ann. Genet. 31, 241–243 (1988).
Chow, C. Y., Zhang, Y., Dowling, J. J., Jin, N., Adamska, M., Shiga, K. et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448, 68–72 (2007).
de Leeuw, C. N. CMT4J: Charcot–Marie–Tooth disorder caused by mutations in FIG4. Clin. Genet. 73, 318–319 (2008).
Ikonomov, O. C., Sbrissa, D., Fligger, J., Delvecchio, K. & Shisheva, A. ArPIKfyve regulates Sac3 protein abundance and turnover: disruption of the mechanism by Sac3I41T mutation causing Charcot–Marie–Tooth 4J disorder. J. Biol. Chem. 285, 26760–26764 (2010).
Acknowledgements
We thank the patient and her family for participating in this work. We also thank Ms S Sugimoto and K Takabe for their technical assistance. This work was supported by research grants from the Ministry of Health, Labour and Welfare (N Matsumoto and N Miyake), the Japan Science and Technology Agency (N Matsumoto), the Strategic Research Program for Brain Sciences (N Matsumoto) and a Grant-in-Aid for Scientific Research on Innovative Areas-(Transcription cycle)-from the Ministry of Education, Culture, Sports, Science and Technology of Japan (N Matsumoto), a Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (H Saitsu, N Matsumoto and N Miyake), the Takeda Science Foundation (N Matsumoto and N Miyake) and the Hayashi Memorial Foundation for Female Natural Scientists (N Miyake).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Nakajima, J., Okamoto, N., Shiraishi, J. et al. Novel FIG4 mutations in Yunis–Varon syndrome. J Hum Genet 58, 822–824 (2013). https://doi.org/10.1038/jhg.2013.104
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.104
Keywords
This article is cited by
-
Severe Consequences of SAC3/FIG4 Phosphatase Deficiency to Phosphoinositides in Patients with Charcot-Marie-Tooth Disease Type-4J
Molecular Neurobiology (2019)
-
Yunis-Varón syndrome caused by biallelic VAC14 mutations
European Journal of Human Genetics (2017)
-
‘Cortical cerebellar atrophy’ dwindles away in the era of next-generation sequencing
Journal of Human Genetics (2014)
