
Abstract
Metachromatic leukodystrophy (MLD) is an autosomal recessive lysosomal storage disease caused by deficiency of the enzyme arylsulfatase A encoded by the ARSA gene located on 22q13.33. Typically, in autosomal recessive disease, a patient inherits two mutations from both parents who are heterozygous carriers. However, in some instances, it is possible to develop the disease by uniparental isodisomy (UPiD), in which two copies of the same mutated allele are inherited from only one carrier parent. Here, we report the first patient with MLD caused by UPiD of chromosome 22. The patient has a homozygous missense mutation, P136T, on ARSA. Family study of the ARSA gene and leukocyte enzyme activity revealed that his father and sister were heterozygous carriers, but his mother possessed only wild-type alleles and normal enzyme activity. Karyotypes of the patient and the parents were normal. Microsatellite analysis showed no discrepancy of parentage, and paternal UPiD of chromosome 22 was indicated. Finally, genome-wide single-nucleotide polymorphism array confirmed the region of UPiD was extended to the entire chromosome 22 of the patient.
Similar content being viewed by others

Main
Metachromatic leukodystrophy, MLD (MIM no. 250100), is an autosomal recessive (AR) inherited demyelinating storage disease caused by deficient activity of the lysosomal enzyme arylsulfatase A encoded by the ARSA gene (MIM no. 607574) located on 22q13.33. Clinically, there are three major subtypes, late-infantile, juvenile and adult MLD, classified by age of onset and severity. Usually, in an autosomal recessive disease, both parents of the patient are heterozygous carriers of the mutation, and offspring have a 25% risk of homozygous mutation and disease development. However, in some instances, a single mutated allele from only one parent will lead to disease due to uniparental disomy (UPD). UPD is defined as the inheritance of a pair of homologous chromosomes from only one parent, both homologous (heterodisomy) or two copies of one homologous (isodisomy). To date, various AR diseases caused by uniparental isodisomy (UPiD) were known.1, 2, 3 In this study, we report a boy with late-infantile MLD caused by paternal UPiD of the entire chromosome 22, confirmed by genome-wide single-nucleotide polymorphism (SNP) array technology. To our knowledge, this is the first patient with AR disease caused by UPD 22.
The patient was born to non-consanguineous Japanese parents at 39 weeks gestational age. During the pregnancy, routine fetal ultrasonograhy detected no abnormality of the fetus and the placenta. His birth weight was 3176 g (+0.4 s.d.), length 48.0 cm (−0.5 s.d.) and head circumference 36.0 cm (+1.9 s.d.). His physical condition and appearance, also the size and macroscopic investigation of the placenta were normal. After birth, his growth were normal, but his psychomotor development was slightly delayed. He gained head control, sitting, crawling, standing and walking at the age of 4, 8, 10, 14 and 16 months, respectively. He started to speak a few words at 1 year. His gait remained unstable, and after he developed exanthema subitum at 1 year and 11 months of age, he could no longer walk alone. He visited our hospital at 2 years of age. He has a 4-year-old healthy sister. He had no obvious facial, limb, genital or body malformations. Pyramidal signs in the lower extremities became obvious and brain magnetic resonance imaging imaging at 2 years and 2 months of age showed leukodystrophy of deep white matter typical of MLD (Figure 1).4 Reduced arylsulfatase A activity in leukocytes of the patient confirmed the diagnosis. Interestingly, the father and the sister had an ∼50% reduction in enzyme activity and were suspected carriers, however, the mother had normal enzyme activity (Figure 2). To investigate further details, we performed molecular analysis. All study participants provided informed consent, and the study design was approved by an ethics review board of Kanazawa University.

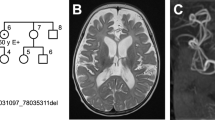
Brain MRI (magnetic resonance imaging) image of the patient. T2 weighted image of (a) axial, (b) coronal and (c) sagittal sections. Massive leukodystrophy of deep white mater and a relatively preserved perivascular area create a typical mottled appearance of metachromatic leukodystrophy (MLD), referred to as leopard skin or tigroid pattern.

Molecular and enzymatic analysis of the family. ARSA mutation analysis and microsatellite analysis of chromosome 22 marker showed paternal uniparental isodisomy of the proband.
Karyotypes of the patient and parents were normal. Entire coding exons of the ARSA gene were amplified by PCR from peripheral blood DNA using originally designed primer sets (Supplementary Table 1) and the resultant products were subjected to bidirectional sequencing. ARSA gene analysis showed a P138T homozygous mutation in the patient. This mutation has not been reported previously, but two missense mutations of the same codon, P138S and P138L were reported.5, 6 Two web-based algorithmic programs, SIFT7 and PMut8 predict that this substitution is pathological. We also checked 100 Japanese normal volunteers DNA and P138T did not appear. According to these results, we concluded that P138T is a disease causative mutation. P138 is one of the evolutionarily conserved amino-acid residue identical in several other human alylsulfatase family and in arylsulfatase from other species.5 Substitution of this amino-acid residue is expected to affect the protein function profoundly and correlate to the severe phenotype. Accordingly, our patient with P138T homozygous mutation was late-infantile type. Also, previously reported MLD patient with P138S/T329I compound heterozygous mutation showed marked reduction of ARSA enzyme activity and classified as late-infantile type.5 ARSA gene analysis of the family revealed that the father and the sister are carriers, but the mother has no mutated allele (Figure 2).
To verify the parentage and segregation of chromosome 22, microsatellite analysis was performed. Polymorphic markers were amplified by PCR and run on polyacrylamide gel then developed with a silver stain. Observed polymorphisms for each microsatellite marker were assigned a numerical designation based on the relative size of the PCR products from the smallest to the largest. Of the 13 microsatellite markers other than chromosome 22, 10 markers were informative and the parentage is consistent. For the four chromosome 22 markers, all markers were informative, and D22S929 and D22S430 showed no maternal alleles and only one paternal allele was inherited by the patient (Figure 2). These results strongly suggested paternal UPiD of chromosome 22. To confirm and extend the results, microarray analysis of the patient was performed using an Affymetrix genome-wide human SNP 6.0 array (Affymetrix Inc., Santa Clara, CA, USA). This array detects the region of UPiD as a copy number neutral loss of heterozygosity (LOH) by the analysis of vast amounts of SNP genotyping. In the chromosomal region belonging to UPiD, only the homozygous SNP alleles (AA or BB) but heterozygous SNP alleles (AB) are present, with two copies of DNA consistently throughout the region. SNP array clearly showed UPiD of the patient extended to the entirety of chromosome 22 (Figure 3). Comparing the SNP allele calls between the patient and parents confirmed paternal UPiD of chromosome 22. Within the 11 471 SNP probes on chromosome 22, the genotype of 1471 SNPs (10.3%) in the patient were informative for parental origin and all of them showed only paternal allele were inherited by the patient. In this 1471 SNPs, the genotype of the 839 SNPs were informative as a marker for paternal heterozygote, and the all genotype of the 839 SNPs were homozygote of one of the paternal alleles in the patient, demonstrating the paternal isodisomy (Supplementary Table 2). Also, copy number state of the patient’s array data showed two copies of DNA was maintained around the ARSA locus (Supplementary Table 3). According to SNP array results, karyotype of the patient is represented as upd(22)pat.arr 22q11.21q13.33(16 055 171–51 234 443) × 2 hmz.

Results from the SNP array analysis of the patient illustrated by the Affymetrix Genotyping Console browser. Uniparental isodisomy (UPiD) spanning the entire chromosome 22 is indicated as a copy number neutral LOH region (boxed region). This region demonstrates the absence of AB genotypes, as seen in the allele difference track, and two copy number state throughout the region. The ARSA gene is located at 22q13.3, within the region of LOH, as shown by the vertical line demarcated with an arrow. Data analysis was performed using Genotyping Console v4.1.1, along with the BIRDSEED V2 algorithm to determine genotyping calls (Affymetrix Inc.). Affymetrix genome-wide human single-nucleotide polymorphism (SNP) 6.0 array mounts 946 000 non-SNP copy number detection probes and 906 600 SNP probes for the entire genome, including 11 400 SNP probes for chromosome 22.
The concept of UPD was first reported over 30 years ago.9 After that, UPD of nearly all chromosomes has been reported.3 When the chromosome involved in UPD contains imprinting genes, overexpression or loss of expression of these genes could cause disease. Conversely, when the UPD chromosome does not contain any imprinting genes, UPD does not readily affect the phenotype. For chromosome 22, both paternal and maternal UPD seemed to not affect the phenotype. In one family, the father had a robertsonian translocation of chromosome 22; 45,XY,der(22;22)(q10;q10) and the son’s karyotype was 45,XY,der(22;22)(q10;q10)pat. The phenotype of the son is completely normal including intelligence and growth.10 Similarly, de novo 45,XY,der(22;22)(q10;q10) was detected in a normal healthy male during the investigation of his wife’s repetitive spontaneous abortions, and microsatellite analysis proved he inherited only one allele of maternal chromosome 22.11 In our study, the patient had no obvious malformations and growth failure, and the clinical course was not distinguished from usual late-infantile MLD. Several mechanisms leading to UPD are estimated,12 which include monosomy rescue, post-fertilization error (mitotic non-disjunction with duplication or mitotic recombination), trisomy rescue after non-disjunction of meiosis I or II and gamete complementation (fertilization of disomic and nullisomic gametes). Of which, gamete complementation, non-disjunction of meiosis I and events, including homologous recombination, are resulted in uniparental heterodisomy. Therefore, like the patient of in this study, complete isodisomy of whole chromosome is possibly generated by monosomy rescue, mitotic non-disjunction with duplication or trisomy rescue after non-disjunction of meiosis II without chiasma formation of the chromosome. In monosomy rescue, mosaicism is unlikely because of the lethality of the monosomic cell line. On the other hand, trisomy rescue could be accompanied with mosaicism. In our patient, 50 metaphases of G-banding karyotypes were analyzed and no existence of mosaicism was detected. Unfortunately, histological and/or molecular analysis of the placenta was not performed because of normal pregnant course and delivery.
Until recently, UPD was proven by microsatellite analysis. This method is easy and cost effective, but has the disadvantage that a single marker determines a spot information of a chromosome, and a marker is not always informative to confirm UPiD. This weakness makes it difficult to decide the accurate site and range of UPD. SNP array technology overcomes this problem using vast amounts of SNP markers along the genome, and permits site accuracy and determination of the extent of UPD even in a cases of partial UPiD created by recombination of homologous chromosomes during meiosis.13 Determination of UPD in a family with autosomal recessive disease is clinically important especially for genetic counseling. Hence, when a proband is affected by UPiD, the recurrence risk for the patient’s sibling is extremely low.12 SNP array technology allows more accurate and comprehensive detection of UPD compared with microsatellite analysis and is expected to become mainstream.
References
Nimmo, G., Monsonego, S., Descartes, M., Franklin, J., Steinberg, S. & Braverman, N. Rhizomelic chrondrodysplasia punctata type 2 resulting from paternal isodisomy of chromosome 1. Am. J. Med. Genet. A 152A, 1812–1817 (2010).
Turner, C. L., Bunyan, D. J., Thomas, N. S., Mackay, D. J., Jones, H. P., Waterham, H. R. et al. Zellweger syndrome resulting from maternal isodisomy of chromosome 1. Am. J. Med. Genet. A 143A, 2172–2177 (2007).
Zlotogora, J. Parents of children with autosomal recessive diseases are not always carriers of the respective mutant alleles. Hum. Genet. 114, 521–526 (2004).
Cheon, J. E., Kim, I. O., Hwang, Y. S., Kim, K. J., Wang, K. C., Cho, B. K. et al. Leukodystrophy in children: a pictorial review of MR imaging features. Radiographics 22, 461–476 (2002).
Gort, L., Coll, M. J. & Chabás, A. Identification of 12 novel mutations and two new polymorphisms in the arylsulfatase A gene: haplotype and genotype-phenotype correlation studies in Spanish metachromatic leukodystrophy patients. Hum. Mutat. 14, 240–248 (1999).
Gieselmann, V., Polten, A., Kreysing, J. & von Figura, K. Molecular genetics of metachromatic leukodystrophy. J Inherit. Metab. Dis. 17, 500–509 (1994).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Ferrer-Costa, C., Gelpí, J. L., Zamakola, L., Parraga, I., de la Cruz, X. & Orozco, M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 21, 3176–3178 (2005).
Engel, E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am. J. Med. Genet. 6, 137–143 (1980).
Ouldim, K., Sbiti, A., Natiq, A., El-Kerch, F., Cherkaoui, S. & Sefiani, A. Unexpected fertility and paternal UPD 22. Fertil. Steril. 90, e13–e15 (2008).
Schinzel, A. A., Basaran, S., Bernasconi, F., Karaman, B., Yüksel-Apak, M. & Robinson, W. P. Maternal uniparental disomy 22 has no impact on the phenotype. Am. J. Hum. Genet. 54, 21–24 (1994).
Yamazawa, K., Ogata, T. & Ferguson-Smith, A. C. Uniparental disomy and human disease: an overview. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 329–334 (2010).
Cottrell, C., Mendell, J., Hart-Kothari, M., Ell, D., Thrush, D., Astbury, C. et al. Maternal uniparental disomy of chromosome 4 in a patient with limb-girdle muscular dystrophy 2E confirmed by SNP array technology. Clin. Genet. 81, 578–583 (2012).
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and a grant from the Ministry of Health, Labor and Welfare of Japan, Tokyo.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Niida, Y., Kuroda, M., Mitani, Y. et al. Paternal uniparental isodisomy of chromosome 22 in a patient with metachromatic leukodystrophy. J Hum Genet 57, 687–690 (2012). https://doi.org/10.1038/jhg.2012.97
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.97
Keywords
This article is cited by
-
Late infantile metachromatic leukodystrophy: Clinical manifestations of five Taiwanese patients and Genetic features in Asia
Orphanet Journal of Rare Diseases (2015)
-
Maternal Uniparental Isodisomy Causing Autosomal Recessive GM1 Gangliosidosis: A Clinical Report
Journal of Genetic Counseling (2014)
