
Abstract
Genomic imprinting is an important epigenetic phenomenon in studying complex traits and has generally been examined by detecting parent-of-origin effects of alleles. The parental-asymmetry test (PAT) based on nuclear families with both parents and its extensions to deal with missing parental genotypes is simple and powerful for such a task. However, these methods only use case (affected) children in nuclear families and thus do not make full use of information on control (unaffected) children, if available, in these families. In this article, we propose a novel parent-of-origin effects test C-PATu (the combined test of PATu and 1-PATu) by using both the control and case children in nuclear families with one or both parents. C-PATu is essentially a weighted framework, in which the test based on all the control children and their parents and that based on all the case children and their parents are weighted according to the population disease prevalence. Simulation results demonstrate that the proposed tests control the size well under no parent-of-origin effects and using additional information from control children improves the power of the tests under the imprinting alternative. Application of C-PATu to a Framingham Heart Study data set further shows the feasibility in practical application of the test.
Similar content being viewed by others

Introduction
Genomic imprinting is an important epigenetic factor in studying complex traits, where the expression level of a gene depends on whether it is paternally or maternally inherited. An imprinted gene database was constructed,1 and 72 imprinted genes in human have been reported up to date (http://igc.otago.ac.nz). For some complex diseases, such as Beckwith–Wiedemann, Prader–Willi and Angelman syndromes, diabetes and schizophrenia, imprinting effects have been demonstrated or hypothesized to have an important role.2, 3, 4, 5, 6, 7, 8, 9, 10 Further, incorporating information on imprinting effects into association analysis could improve the test power.11 On the other hand, genomic imprinting has been largely examined in analyses of complex traits through testing for parent-of-origin effects of alleles.12 For a diallelic marker locus (ML), the parental-asymmetry test (PAT) based on nuclear families with both parents13 and its extensions to deal with missing parental genotypes14 is simple and powerful for such a task, in the absence of maternally mediated genetic effects. However, these methods only use case (affected) children in nuclear families and thus do not make full use of information on control (unaffected) children, if available, in these families.
Recently, there has been a considerable interest in the incorporation of information on control children in nuclear families into association study and detection of parent-of-origin effects. A hybrid design was proposed to augment a set of affected individuals and their parents with a set of unaffected, unrelated individuals and their parents, which can provide more power than either the case–parent approach or the case–mother/control–mother approach for detecting the association.15, 16 On the other hand, a multinomial modelling approach was proposed to investigate effects of parent-of-origin, maternal genotype and maternal–fetal genotype interactions using either case/mother duos or case/parent trios, which could incorporate additional types of control sample (such as unrelated controls, controls and their mothers or both parents of controls) into analysis.17 However, it is only suitable for families with a single child, irrespective of affected or unaffected. Yang and Lin18 developed a likelihood approach for detecting imprinting and maternal effects (LIME) using nuclear families with multiple affected and unaffected children from a prospective study, while they found that the above-mentioned model-free methods13, 14 are more powerful than LIME when there is no maternal effect.
In this article, for nuclear families with both parents available, we firstly propose a novel PAT0 by only using control children and their parents in the presence of association between an allele at the ML under study and a disease gene. For families with only one parent available, we develop a 1−PAT0 test to test for parent-of-origin effects based on all the control–parent pairs in the sample. For a mixture of families with both parents and families with only one parent, C-PAT0 (the combined test of PAT0 and 1−PAT0) is suggested. Then, by using both affected and unaffected children in the sample, C-PATu is proposed. C-PATu is essentially a weighted framework, in which the test based on all the control children and their parents and the test based on all the case children and their parents are weighted according to the population disease prevalence. Simulation results demonstrate that the proposed tests control the size well under the null hypothesis of no parent-of-origin effects and are robust to population stratification. We compare C-PATu with the existing method that omits unaffected children in its analysis under four family samples, four types of parent-of-origin effects models, various missing father rates and incomplete family rates. The results demonstrate that using additional information from the control children in nuclear families improves the power of the tests. Application of the proposed C-PATu test to a Framingham Heart Study data set further shows the feasibility in practical application of the test.
Materials and methods
Background and notations
Suppose that a ML of interest has two alleles M1 and M2. For convenience, let 0, 1 and 2, respectively, represent the marker genotypes M2M2, M1M2 and M1M1, which are the number of copies of allele M1 in each genotype. For a child–parents trio, let F, M and C denote the genotypes of the father, mother and child, respectively, and then F, M and C take possible values of 0, 1 or 2. Consider a disease susceptibility locus (DSL) with mutant allele D and normal allele d. The four ordered genotypes at the DSL are D/D, D/d, d/D and d/d, and the corresponding genotype frequencies are P(D/D), P(D/d), P(d/D) and P(d/d), respectively. Without loss of generality, we assume that the left allele of the slash is paternal and the right one is maternal. Let ϕD/D, ϕD/d, ϕd/D and ϕd/d be the penetrance of genotypes D/D, D/d, d/D and d/d, respectively. Thus, the degree of imprinting I=(ϕD/d−ϕd/D)/2 is used to measure parent-of-origin effects. I=0 suggests no parent-of-origin effects. There are two extreme cases, one is complete paternal parent-of-origin effect (ϕD/d=ϕd/d and ϕd/D=ϕD/D) and the other is complete maternal parent-of-origin effect (ϕD/d=ϕD/D and ϕd/D=ϕd/d). Let A be the disease indicator for a child. That is, we assume that ‘A=0’ and ‘A=1’ denote the events that the child is unaffected and affected, respectively. Then, the population disease prevalence is c=ϕD/DP(D/D)+ϕD/dP(D/d)+ϕd/DP(d/D)+ϕd/dP(d/d).
Note that there are four haplotypes M1D, M1d, M2D and M2d at the ML and DSL. Let θ be the recombination fraction between the ML and DSL. To test for parent-of-origin effects, we assume that the ML and DSL are in linkage disequilibrium. Just like the literature,13, 14 mating symmetry is assumed throughout this article, that is, P(F=f, M=m)=P(F=m, M=f) for all f, m=0, 1, 2. We further assume that there are no maternally mediated genetic effects.
Methods only using unaffected children and their parents
Firstly, we consider the situation where all the families are control–parents trio with known marker genotypes of an unaffected (control) child and his/her parents. For control–parents trios, all the genetically possible 15 types of family and the conditional probabilities (given that the child is a control) t1,t2,…,t15 are listed in Table 1, whereas the corresponding conditional probabilities for case–parents trios are termed s1,s2,…,s15, respectively. It is shown in Supplementary Information that t3=t4, t8=t9 and t13=t14 under the null hypothesis of no parent-of-origin effects. So, for nu independent control–parents trios,  which is zero under the null. Here, Fi, Mi and Ci are the genotypes of the father, mother and unaffected child in the ith family, respectively, i=1,…,nu; I(comparison statement) is 1 if the comparison statement holds and is 0 otherwise. Further, we could obtain that
which is zero under the null. Here, Fi, Mi and Ci are the genotypes of the father, mother and unaffected child in the ith family, respectively, i=1,…,nu; I(comparison statement) is 1 if the comparison statement holds and is 0 otherwise. Further, we could obtain that  is an unbiased estimate of variance of
is an unbiased estimate of variance of  under the null hypothesis (see Supplementary Information). Therefore, we propose PAT0 to detect parent-of-origin effects for control–parents trios as
under the null hypothesis (see Supplementary Information). Therefore, we propose PAT0 to detect parent-of-origin effects for control–parents trios as  which asymptotically follows a standard normal distribution under the null.
which asymptotically follows a standard normal distribution under the null.
Now, we extend the PAT0 to accommodate more general families with multiple unaffected children (proof omitted for brevity). Consider n independent nuclear families, each with known marker genotypes for the father, mother and children, and the known affection statuses of the children (affected or unaffected). Suppose that Fi and Mi are the genotypes for the father and mother in the ith family, respectively, i=1,2,…,n. Let n0i and n1i be the numbers of control and case children in the ith family, respectively. C0ij denotes the genotype of the jth control child in the ith family, i=1,2,…,n, j=1,2,…,n0i, and C1ij represents the genotype of the jth case child in the ith family, i=1,2,…,n, j=1,2,…,n1i. For each nuclear family, let every unaffected child be matched with both parents and the resulting trio is naturally termed as a control–parents trio; let every affected child be matched with both parents and the resulting trio is termed as a case–parents trio. Based on all the control–parents trios in the sample,  is proposed to detect parent-of-origin effects, where
is proposed to detect parent-of-origin effects, where


 denotes the total number of paired unaffected children who are heterozygous in family i in which the parents have different marker genotypes. The first subscript 0 in X02 and Y02 denotes the case of only using unaffected children (A=0), and the second subscript 2 represents the case based on families with both parents available (complete nuclear families).
denotes the total number of paired unaffected children who are heterozygous in family i in which the parents have different marker genotypes. The first subscript 0 in X02 and Y02 denotes the case of only using unaffected children (A=0), and the second subscript 2 represents the case based on families with both parents available (complete nuclear families).
Note that the incorporation of incomplete nuclear families (families in which not both parents are available) could improve the statistical power of a test. However, families with no parental genotypes are not informative for imprinting.10, 14 The incomplete nuclear families hereafter simply refer to those with only one parent. Suppose that we have nM incomplete nuclear families in which the fathers are missing (mothers are available) and nF incomplete nuclear families in which the mothers are missing (fathers are available). So, we have a total of nI=nM+nF incomplete nuclear families. For each incomplete nuclear family, let every child be paired with the available parent and the resulting pair is termed as a child–parent pair. If there are n0i unaffected children in the ith incomplete family, 1⩽i⩽nI, then we have  control–mother pairs and
control–mother pairs and  control–father pairs. If there are n1i affected children in the ith incomplete family, 1⩽i⩽nI, then we have
control–father pairs. If there are n1i affected children in the ith incomplete family, 1⩽i⩽nI, then we have  case–mother pairs and
case–mother pairs and  case–father pairs. Based on all the control–parent pairs in the sample, we propose the statistic
case–father pairs. Based on all the control–parent pairs in the sample, we propose the statistic  where
where


and w0=n0CF/(n0CM+n0CF). The first subscript 0 in X01 and Y01 denotes the case of all the involved children being unaffected (A=0) and the second subscript 1 represents the case based on families with only one parent available. It is shown in Supplementary Information that 1−PAT0 is valid in detecting parent-of-origin effects.
When we collect n complete nuclear families, nM incomplete nuclear families with missing father and nF incomplete nuclear families with missing mother, the combined test statistic C-PAT0 of PAT0 and 1−PAT0 is proposed to test for parent-of-origin effects as  where X02 and Y02 are defined in Equations (1) and (2), and X01 and Y01 are defined in Equations (3) and (4), respectively.
where X02 and Y02 are defined in Equations (1) and (2), and X01 and Y01 are defined in Equations (3) and (4), respectively.
Methods using both unaffected and affected children and their parents
Note that the methods PAT, 1−PAT and C-PAT proposed by Zhou et al.14 are suitable for all the case–parents trios and/or case–parent pairs in the sample. To make full use of available information on unaffected and affected children in the sample, we combine these methods with PAT0, 1−PAT0 and C-PAT0 in the following, respectively. Further, it is shown in Supplementary Information that [(s3−s4)+(s8−s9)+(s13−s14)]:[(t4−t3)+(t9−t8)+(t14−t13)]=(1−c):c. So, we assign the weights 1−c and c to the contribution from affected and unaffected children, and propose the following three combined statistics PATu, 1−PATu and C-PATu to test for parent-of-origin effects by incorporating unaffected children


where the detailed expressions of X12, Y12, X11 and Y11 for case children and their parents refer to Supplementary Information, and the first subscript 1 denotes the case of only using affected children (A=1) and the second subscript 2 (1) represents the case based on families with both parents (only one parent) available. Further,


PATu, 1−PATu and C-PATu have the approximate standard normal distributions under the null (see Supplementary Information). Note that these three statistics use the following three types of nuclear families (irrespective of both parents or only one parent available): (1) nuclear families in which all the children are affected; (2) nuclear families in which all the children are unaffected and (3) nuclear families in which some children are affected and some are unaffected. As such, PATu, 1−PATu and C-PATu take account of dependencies among all the children in each family, irrespective of unaffected or affected.
It should be noted that the population disease prevalence c is included in C-PATu and then is generally presumed to be known prior to analysis. If it is unknown, then it is estimated by the ratio of the number of affected children to the total number of children in the sample.19 This estimate may be biased because of some reasons, such as the ascertainment scheme that a family to be included should have at least one affected child. However, this will not affect the validity of the proposed methods but the test power (see Results).
Results
Settings
In this section, simulations are conducted to check the validity of PATu, 1−PATu and C-PATu, and to make the power comparison of PATu with PAT, 1−PATu with 1−PAT and C-PATu with C-PAT. Consider the population stratification demographic model, which comprises two different homogeneous subpopulations in the population under study. A family came from the first (second) population with probability 0.7 (0.3) and the frequencies of haplotypes M1D, M1d, M2D and M2d in the first (second) population are taken to be 0.2, 0.15, 0.05 and 0.6 (0.4, 0.1, 0.1 and 0.4), respectively. The recombination fraction between the ML and DSL is fixed at θ=0.001.
To study the size of all the proposed methods, we consider simulations under a model without parent-of-origin effects (ϕD/D=0.6, ϕD/d=ϕd/D=0.4 and ϕd/d=0.2). Four parent-of-origin effects models are utilized in the power comparison of PATu with PAT: complete paternal parent-of-origin effect (PEM1: ϕD/D=ϕd/D=0.6 and ϕD/d=ϕd/d=0.2), incomplete paternal parent-of-origin effect (PEM2: ϕD/D=0.6, ϕD/d=0.25, ϕd/D=0.55 and ϕd/d=0.2; PEM3: ϕD/D=0.6, ϕD/d=0.3, ϕd/D=0.5 and ϕd/d=0.2; PEM4: ϕD/D=0.6, ϕD/d=0.35, ϕd/D=0.45 and ϕd/d=0.2), which corresponds to the degree of imprinting I taking values from −0.20 to −0.05 in increments of 0.05. However, for simplicity, we only consider the models PEM1 and PEM2 in the simulations for 1−PATu/1−PAT and C-PATu/C-PAT.
We use four types of family samples: FS1: 200 families, each with three offspring, of which at least one is affected, and 100 control–parents trios; FS2: 200 families, each with three offspring, of which at least one is affected; FS3: 100 families, each with three offspring, of which at least one is affected, and 100 control–parents trios; and FS4: 100 families, each with three offspring, of which at least one is affected. For each subpopulation, parental haplotypes at the ML and a DSL are generated according to those four haplotype frequencies. Then, the haplotypes of the offspring are generated from their parental haplotypes with the recombination fraction θ. We assign the affection statuses of the offspring based on their genotypes at the DSL and four penetrances ϕD/D, ϕD/d, ϕd/D and ϕd/d. For families with three offsprings, if these families have at least one affected child, then the families are sampled. Otherwise, they are ignored. On the other hand, note that the sum of two heterozygous penetrances is fixed at 0.8. As such, on average, the resulting sample of 200 families with three offspring consists of 121 families with only one affected child, 64 with two affected and 15 with three affected for each parent-of-origin effects model. Similarly, there are approximately 61 families with only one affected child, 32 with two affected and 7 with three affected in the sample of 100 families with three offspring.
For extensive assessment of the proposed tests (PATu, 1−PATu and C-PATu) and for comparison with PAT, 1−PAT and C-PAT, we consider the following statistics. Two PATu (1−PATu or C-PATu) statistics are based on family sample FS1 for the population (true) disease prevalence c and the estimated c value, which are denoted as PATu1T (1−PATu1T or C-PATu1T) and PATu1E (1−PATu1E or C-PATu1E), respectively. Similarly, we obtain the corresponding notations for PATu, 1−PATu and C-PATu based on FS2, FS3 and FS4 for the true and estimated c values (omitted here for brevity). Note that PAT, 1−PAT and C-PAT only utilize the case children in the samples. So, we consider families with at least one affected child and denote PAT, 1−PAT and C-PAT based on 200 families each with three children, of which at least one is affected (FS2) as PAT2, 1−PAT2 and C-PAT2, and those based on 100 families, each with three children, of which at least one is affected (FS4) as PAT4, 1−PAT4 and C-PAT4, respectively.
When there are missing data on parental genotypes, the incomplete family rate τ is used to determine whether a family is incomplete (only one parent is available) or complete (both parental genotypes are available). The incomplete family rate ranges from 0 to 1 in increments of 0.1. Further, we use the missing father rate (among the incomplete families) β to determine whether the father or mother is missing in an incomplete family. In the simulations for 1−PATu/1−PAT, β takes values from 0.2 to 0.8 in increments of 0.1, whereas the simulations for C-PATu/C-PAT are all performed with β=0.5 for brevity. We evaluate the actual size and power based on 10000 replicates and at significance level α=5%. Other additional simulations with less genotype relative risks are conducted and the results are shown in Supplementary Table 1.
Size and power of PATu and PAT
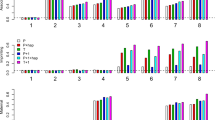
Figure 1 displays the size and power of PATu (based on the population disease prevalence and the estimated prevalence) for family samples FS1–FS4 and PAT for FS2 and FS4 against the degree of imprinting I. It is shown in the figure that the actual size of PATu stays at the nominal 5% level (I=0), regardless of the population/estimated disease prevalence or the types of family samples, signifying the validity of PATu. Further, the PATu by picking up the information on control children in the sample is indeed more powerful than PAT excluding all the control children (I≠0). It should also be noted that the power of PATu for FS1 and FS3 consisting of 100 additional control–parents trios on the basis of the population prevalence c and that on the basis of the estimated c value are almost the same (PATu1T vs PATu1E in Figure 1a and PATu3T vs PATu3E in Figure 1b). However, for FS2 and FS4, the PATu based on the estimated c value has less power than that based on the population prevalence, because of the bias of the estimate of c caused by the ascertainment scheme that the families with at least one affected child are included in the sample (PATu2T vs PATu2E in Figure 1a and PATu4T vs PATu4E in Figure 1b). By comparing the power of PATu for FS1 (FS3) with that for FS2 (FS4), we find that incorporating additional control–parents trios can improve the test power (PATu1T vs PATu2T or PATu1E vs PATu2E in Figure 1a and PATu3T vs PATu4T or PATu3E vs PATu4E in Figure 1b). Finally, the figure shows that the power of PATu and PAT increases when |I| increases and the power under FS1 (FS2) with larger sample size is larger than that under FS3 (FS4) with smaller sample size (Figure 1a vs b).

Power comparison of PATu and PAT against degree of imprinting I based on 10000 replicates. (a) PATu1T and PATu1E are based on family sample FS1 (200 families with at least one affected child and 100 control–parents trios) for the true disease prevalence c and the estimated c value, respectively; PATu2T for FS2 (200 families with at least one affected child) and true c value; PATu2E for FS2 and estimated c value; PAT2 for FS2. (b) PATu3T for FS3 (100 families with at least one affected child and 100 control–parents trios) and true c value; PATu3E for FS3 and estimated c value; PATu4T for FS4 (100 families with at least one affected child) and true c value; PATu4E for FS4 and estimated c value; PAT4 for FS4.
Size and power of 1−PATu and 1−PAT
Table 2 gives the actual size of 1−PATu for family samples FS1–FS4 based on the population disease prevalence and the estimated c value against missing father rates. It can be seen that the test is slightly conservative, irrespective of the population/estimated prevalences, missing father rates or family sample types.
Figure 2 plots the power of 1−PATu against different missing father rates β under family samples FS1–FS4, when I takes values of −0.20 and −0.15. The effect of β on the power of 1−PATu cannot be ignored and the maximum power occurs when β=0.5. From the figure, we also find that the power of 1−PATu by incorporating all the additional control–parent pairs into analysis is larger than 1−PAT only using case–parent pairs, and the 1−PATu based on the estimated c value is less powerful than that based on the population prevalence under the ascertainment scheme with at least one affected child in each family. These findings are consistent with our observation from Figure 1. Again, as shown in Figure 2, there is a greater power for detecting parent-of-origin effects when |I| becomes larger (Figure 2a vs b or c vs d).

Power comparison of 1−PATu and 1−PAT against missing father rate β based on 10000 replicates. Top two subplots (a) I=−0.20 and (b) I=−0.15: 1−PATu1T and 1−PATu1E are based on family sample FS1 (200 families with at least one affected child and 100 control–parents trios) for the true disease prevalence c and the estimated c value, respectively; 1−PATu2T for FS2 (200 families with at least one affected child) and true c value; 1−PATu2E for FS2 and estimated c value; 1−PAT2 for FS2. Bottom two subplots (c) I=−0.20 and (d) I=−0.15: 1−PATu3T for FS3 (100 families with at least one affected child and 100 control–parents trios) and true c value; 1−PATu3E for FS3 and estimated c value; 1−PATu4T for FS4 (100 families with at least one affected child) and true c value; 1−PATu4E for FS4 and estimated c value; 1−PAT4 for FS4.
Size and power of C-PATu and C-PAT
Table 3 shows the size of C-PATu for family samples FS1–FS4 and different incomplete family rates τ, having β=0.5. The empirical size all maintains close to the nominal 5% level, which signifies the validity of C-PATu. Figure 3 shows the power of C-PATu against different incomplete family rates under family samples FS1 and FS2 (the results for FS3 and FS4 omitted for brevity), when I takes values of −0.20 and −0.15 and β is fixed at 0.5. It is shown in Figure 3 that C-PATu by using both the unaffected and affected children in the sample is more powerful than C-PAT only using affected children. The figure also shows that the power of C-PATu increases when |I| increases (Figure 3a vs b). The power of C-PATu generally decreases with τ increasing. However, it should be noted from the figure that C-PATu is somewhat less powerful when τ=90% than when τ=100%. This could be because the heterogeneous information on the mixture of both complete and incomplete families (τ=0.9) may cause a higher variability than the homogeneous information on only incomplete families (τ=1).

Power comparison of C-PATu and C-PAT against incomplete family rate τ based on 10000 replicates, with β=0.5. C-PATu1T and C-PATu1E are based on family sample FS1 (200 families with at least one affected child and 100 control–parents trios) for the true disease prevalence c and the estimated c value, respectively; C-PATu2T for FS2 (200 families with at least one affected child) and true c value; C-PATu2E for FS2 and estimated c value; C-PAT2 for FS2. (a) I=−0.20; (b) I=−0.15.
Application to Framingham Heart Study data
We applied C-PATu to the Framingham Heart Study data, which were made available through the database of Genotypes and Phenotypes supplied by the Genetic Analysis Workshop 16 (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000128.v3.p3). This data set consists of 1538 pedigrees and 15876 individuals (6210 founders and 9666 nonfounders). However, there are only about 6849 individuals genotyped at 48071 single-nucleotide polymorphism (SNP) markers over the 22 autosomes.
High blood pressure in an adult is defined as a blood pressure ⩾140 mm Hg systolic blood pressure or ⩾90 mm Hg diastolic blood pressure (DBP). In the Framingham Heart Study, the systolic blood pressure and DBP of the original cohort (the first generation) and their offspring (the second generation) were measured in four exams and once in the third generation. Based on the highest measurements among all the available ones for each individual, there were 1733 individuals having high blood pressure among 6845 individuals with at least one systolic blood pressure/DBP measurement in the data set. Further, Egan et al.20 reported the 28.5% rate of hypertension in the US adult population, which is taken as the population prevalence for systolic blood pressure and DBP in this real data application.
We only focus on the analysis for DBP in the following for brevity. Specifically, we use C-PATu to detect parent-of-origin effects in the presence of association between marker genes and DBP. So, we first use the pedigree disequilibrium test21 to identify SNPs that are associated with DBP and then carry out parent-of-origin effects tests only at those identified SNPs. On the other hand, C-PATu can only tackle two-generation nuclear families and then we need to cut each three-generation pedigree into multiple two-generation nuclear families. We randomly select one nuclear family from each three-generation pedigree to be included in the analysis, except for the following constrained conditions: (1) families with both parental genotypes missing were omitted from our analysis, because they are not informative for imprinting and (2) families with at least one affected child are preferentially chosen. This selection process led to 604 nuclear families (comprising 171 families with at least one affected child and 433 families in which all the children are unaffected) with the number of unaffected/affected children in each family ranging 1–7 for each SNP. Our analysis is based on these phenotypic data and the genotypes of these 604 nuclear families.
Totally, 208 SNPs were identified by the pedigree disequilibrium test at the 0.5% significance level. Note that we regarded the association analysis as a preliminary step. As such, we used a less stringent criterion for reducing the chance of missing any potential SNPs in the subsequent parent-of-origin effects test. C-PATu identified five SNPs at the significance level of 0.01 (rs4084639 and rs4908446 on chromosome 1, rs2973566 on chromosome 5, rs12773723 and rs1463058 on chromosome 10, with P-values being 0.0018, 0.0051, 0.0049, 0.0034 and 0.0039, respectively). However, the probability that there are five and more significant results for parent-of-origin effects (P<0.01) among the 208 SNPs is 0.059, which is small. So, the five identified SNPs displaying significant parent-of-origin effects are likely to occur by chance. On the other hand, if we further consider multiple testing issue, then all the results on the five SNPs are nonsignificant.
Discussion
In this article, we extend the C-PAT14 to C-PATu by incorporating information on unaffected children from nuclear families to test for parent-of-origin effects in the presence of association. In essence, C-PATu is a weighted framework of the test X02+X01 based on all the unaffected children and the test X12+X11 based on all the affected children (Equation (5)), which are weighted according to the population disease prevalence. Our simulation results show that C-PATu indeed controls the size well under the null hypothesis of no parent-of-origin effects and is robust to population stratification. We further compare C-PATu with C-PAT that omits unaffected children in its analysis under four family samples, four types of parent-of-origin effects models, various missing father rates and incomplete family rates. The results demonstrate that using additional information from unaffected children in nuclear families improves the power of the tests substantially and C-PATu can have an advantage over C-PAT only when one of two heterozygous penetrances is large. Meanwhile, it should be noted that the misspecification of the population prevalence could reduce the power of C-PATu, although this will not affect its validity. Finally, we have successfully applied C-PATu to the Framingham Heart Study data set by using one blood pressure trait (DBP) and shown the feasibility in practical application of the test. Our software, C-PATu, implemented in R (http://www.r-project.org), is freely available at http://www.echobelt.org/web/UploadFiles/CPATu.html, which includes the pedigree disequilibrium test for association, C-PATu and C-PAT for parent-of-origin effects for the easy use.
References
Morison, I. M., Paton, C. J., Cleverley, S. D. The imprinted gene and parent-of-origin effect database. Nucleic Acids Res. 29, 275–276 (2001).
Chatkupt, S., Lucek, P. R., Koenigsberger, M. R., Johnson, W. G. Parental sex effect in spina bifida: a role for genomic imprinting? Am. J. Med. Genet. 44, 508–512 (1992).
Temple, I. K., James, R. S., Crolla, J. A., Sitch, F. L., Jacobs, P. A., Howell, W. M. et al. An imprinted gene(s) for diabetes? Nat. Genet. 9, 110–112 (1995).
van Schothorst, E. M., Jansen, J. C., Bardoel, A. F., van der Mey, A. G., James, M. J., Sobol, H. et al. Confinement of PGL, an imprinted gene causing hereditary paragangliomas, to a 2-cM interval on 11q22-q23 and exclusion of DRD2 and NCAM as candidate genes. Eur. J. Hum. Genet. 4, 267–273 (1996).
Falls, J. G., Pulford, D. J., Wylie, A. A., Jirtle, R. L. Genomic imprinting: implications for human disease. Am. J. Pathol. 154, 635–647 (1999).
Abel, K. M. Fetal origins of schizophrenia: testable hypotheses of genetic and environmental influences. Br. J. Psychiatry 184, 383–385 (2004).
Dong, C. H., Li, W. D., Geller, F., Lei, L., Li, D., Gorlova, O. Y. et al. Possible genomic imprinting of three human obesity-related genetic loci. Am. J. Hum. Genet. 76, 421–437 (2005).
Samaco, R. C., Hogart, A., LaSalle, J. M. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 14, 483–492 (2005).
Ziegler, A., Konig, I. R. A statistical approach to genetic epidemiology: concepts and applications, Wiley-VCh: Weinheim, (2006).
Rampersaud, E., Morris, R. W., Weinberg, C. R., Speer, M. C., Martin, E. R. Power calculations for likelihood ratio tests for offspring genotype risks, maternal effects, and parent-of-origin (POO) effects in the presence of missing parental genotypes when unaffected siblings are available. Genet. Epidemiol. 31, 18–30 (2007).
Xia, F., Zhou, J. Y., Fung, W. K. A powerful approach for association analysis incorporating imprinting effects. Bioinformatics 27, 2571–2577 (2011).
Hager, R., Cheverud, J. M., Wolf, J. B. Maternal effects as the cause of parent-of-origin effects that mimic genomic imprinting. Genetics 178, 1755–1762 (2008).
Weinberg, C. R. Methods for detection of parent-of-origin effects in genetic studies of case-parents triads. Am. J. Hum. Genet. 65, 229–235 (1999).
Zhou, J. Y., Hu, Y. Q., Lin, S., Fung, W. K. Detection of parent-of-origin effects based on complete and incomplete nuclear families with multiple affected children. Hum. Hered. 67, 1–12 (2009).
Weinberg, C. R., Umbach, D. M. A hybrid design for studying genetic influences on risk of diseases with onset early in life. Am. J. Hum. Genet. 77, 627–636 (2005).
Vermeulen, S. H., Shi, M., Weinberg, C. R., Umbach, D. M. A hybrid design: case-parent triads supplemented by control-mother dyads. Genet. Epidemiol. 33, 136–144 (2009).
Ainsworth, H. F., Unwin, J., Jamison, D. L., Cordell, H. J. Investigation of maternal effects, maternal-fetal interactions and parent-of-origin effects (imprinting), using mothers and their offspring. Genet. Epidemiol. 35, 19–45 (2011).
Yang, J., Lin, S. Detection of imprinting and heterogeneous maternal effects on high blood pressure using Framingham Heart Study data. BMC Proc 3, S125 (2009).
Sun, F. Z., Flanders, W. D., Yang, Q. H., Zhao, H. Y. Transmission/disequilibrium tests for quantitative traits. Ann. Hum. Genet. 64, 555–565 (2000).
Egan, B. M., Zhao, Y., Axon, R. N. US trends in prevalence, awareness, treatment, and control of hypertension, 1988-2008. JAMA 303, 2043–2050 (2010).
Martin, E. R., Monks, S. A., Warren, L. L., Kaplan, N. L. A test for linkage and association in general pedigrees: the pedigree disequilibrium test. Am. J. Hum. Genet. 67, 146–154 (2000).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81072386, 11171075), National Basic Research Program of China (2012CB316505) and the Hong Kong RGC GRF Research Grant (HKU 766511M). The Framingham Heart Study research was supported by National Heart, Lung, and Blood Institute contract 2 N01-HC-25195-06 and by a National Heart, Lung, and Blood Institute contract with Affymetrix, Inc for genotyping services (Contract N02-HL-6-4278). The Genetic Analysis Workshop (GAW) was supported by National Institutes of Health grant R01 GM31575. The GAW16 Framingham data used for the analyses described in this article were obtained through Database of Genotypes and Phenotypes (dbGaP numbers 7640-2, 7664-1, and 7325-3). The authors are grateful to the many investigators within the Framingham Heart Study who have collected and managed the data.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Zhou, JY., Mao, WG., Li, DL. et al. A powerful parent-of-origin effects test for qualitative traits incorporating control children in nuclear families. J Hum Genet 57, 500–507 (2012). https://doi.org/10.1038/jhg.2012.58
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.58
Keywords
This article is cited by
-
Generalized disequilibrium test for association in qualitative traits incorporating imprinting effects based on extended pedigrees
BMC Genetics (2017)
-
A powerful association test for qualitative traits incorporating imprinting effects using general pedigree data
Journal of Human Genetics (2015)
-
Detection of imprinting effects for hypertension based on general pedigrees utilizing all affected and unaffected individuals
BMC Proceedings (2014)
-
Nonparametric method for detecting imprinting effect using all members of general pedigrees with missing data
Journal of Human Genetics (2014)
-
Detection of parent-of-origin effects for quantitative traits using general pedigree data
Journal of Genetics (2014)
