
Abstract
Parkinson's disease (PD) is a neurodegenerative disorder characterized by neurodegeneration, most notably of dopaminergic neurons in the substantia nigra. To date, six causative genes have been identified including LRRK2, whose mutations are the most frequent in autosomal dominant PD (Ad-PD). We conducted a comprehensive mutational analysis of LRRK2 in 30 Ad-PD (11 Japanese and 19 Caucasian) families employing a DNA microarray-based resequencing system and direct nucleotide sequence analysis, and identified 23 variants including two known mutations, p.G2019S and p.I1371V, in three Caucasian families and one Caucasian family, respectively, a novel putative pathogenic mutation, p.N1221K, in one Japanese family, and a known nonsynonymous variant, p.G2385R, in two Japanese families. Detailed analysis of the frequency of p.G2385R among 100 Japanese Ad-PD, 73 sporadic PD (sPD) and 238 controls revealed that the frequency of the p.G2385R variant was significantly higher in Ad-PD than in controls (allele frequency, 9.0 vs 2.1%) (χ2=16.32, P=5.34 × 10−5). The p.G2385R variant, however, did not show complete cosegregation with PD. In addition, the frequency of p.G2385R was also higher in sPD than in controls, although not significant (allele frequency, 3.4 vs 2.1%) (χ2=0.76, P=0.38). These observations support the possibility that p.G2385R is associated with an increased risk of PD.
Similar content being viewed by others

Introduction
Parkinson's disease (PD) is a neurodegenerative disorder characterized by rigidity, tremor, akinesia and postural instability. Its pathological features include the progressive degeneration of dopaminergic neurons primarily in the substantia nigra, with Lewy bodies as the pathological hallmark. Although the majority of PD patients have sporadic PD (sPD), various forms of familial PD (FPD) have been recognized. Recent advances in the search for genes causing PD have contributed to a better understanding of the molecular pathophysiology of PD. To date, six genes (SNCA, PARK2, DJ1, PINK1, UCHL1 and LRRK2) have been identified as genes causing FPD.1, 2, 3, 4, 5, 6 The genetic heterogeneity necessitates a comprehensive mutational analysis of these genes for not only molecular diagnosis but also studies of the molecular epidemiology of FPD.
Among the genes causing FPD, LRRK2 is of particular interest, because LRRK2 is by far the most common cause of autosomal dominant PD (Ad-PD). Approximately 4% of Ad-PD patients and 1% of sPD patients have been reported to harbor causative mutations of LRRK2, the most frequent of which is the mutation substituting serine for glycine at codon 2019 (p.G2019S).7 It is mandatory to carry out mutational analysis of LRRK2 to conduct molecular diagnosis and to investigate the molecular epidemiology of Ad-PD. Analysis of the entire 51 exons of LRRK2 by the conventional direct nucleotide sequencing method, however, is very laborious. Therefore, the majority of previous studies have focused on particular exons for mutational analysis,8, 9, 10, 11, 12 making it difficult to obtain accurate data on the molecular epidemiology of Ad-PD caused by LRRK2.
We herein applied a DNA microarray-based resequencing system to the comprehensive mutational analysis of all LRRK2 exons in 30 families with Ad-PD from two cohorts with different ethnic backgrounds.
Materials and methods
Subjects
To directly compare the LRRK2 epidemiologies across ethnic groups in two similarly ascertained cohorts, we conducted a comprehensive resequencing analysis of LRRK2 focusing on 11 Japanese and 19 Caucasian families with Ad-PD employing resequencing DNA microarrays (Table 1a). These families were diagnosed as having Ad-PD when their pedigree members in at least two generations were diagnosed as having PD. Two Caucasian probands were assessed both by the direct sequencing approach and by resequencing microarray analysis.13 To evaluate the molecular epidemiology of LRRK2 variants identified by the resequencing of LRRK2, DNA samples from index patients from 89 Ad-PD families and 73 sPD patients, and samples from 233 Japanese normal controls were further analyzed (Table 1b). All the genomic DNA samples were obtained with the written informed consent of the subjects, and this research project was approved by the Institutional Review Board of the University of Tokyo.
Mutational analysis
The mutational analysis of all 51 LRRK2 exons was accomplished using the DNA microarray-based resequencing system as described elsewhere.14, 15 Briefly, specific PCR was conducted for each exon using previously reported primers.6 After quantification of the PCR products, they were pooled equimolarly, fragmented with DNase I, labeled with biotin, hybridized to microarrays, stained with streptoavidin-phycoerythrin, washed and scanned. Direct nucleotide sequence analysis was also conducted using an automated DNA sequencer and BigDye Terminator ver. 3.1 (Applied Biosystems, Foster City, CA, USA).
Results
Mutational analysis of LRRK2 in 30 Ad-PD families
Twenty-three variants were identified by the resequencing of the 51 exons and the splice sites of LRRK2 in 30 Ad-PD families (Table 2). Of the 10 nonsynonymous variants identified in the 11 Japanese Ad-PD families, two novel variants (p.N1221K and p.N1320S), both of which were identified in one family, respectively, and the previously reported nonsynonymous variant p.G2385R, which was identified in two families, were of interest. Of the 8 nonsynonymous variants identified in the 19 Caucasian Ad-PD families, the previously known mutation p.G2019S, which was identified in three families, and two previously reported nonsynonymous variant p.R1514Q and p.I1371V, both of which were identified in one family, respectively, were of interest. The remaining seven nonsynonymous variants were previously been registered in the dbSNP database (http://www.ncbi.nlm.nih.gov/SNP/index.html) and not considered as mutations causing Ad-PD.
The two novel nonsynonymous variants found in the Japanese Ad-PD families are a variant substituting lysine for asparagine at codon 1221 (p.N1221K) (Figure 1a) and that substituting serine for arginine at codon 1320 (p.N1320S). p.N1221K was located in a highly conserved region (Figure 1b) and not found in 233 controls (466 chromosomes). The index patient with p.N1221K developed levodopa-responsive parkinsonism including asymmetric rigidity and postural instability at age 47. The other novel nonsynonymous variant p.N1320S was found in one of the 11 Japanese Ad-PD families, but was also present in eight controls (allele frequency, 1.7%). In addition, the variant was also found in three additional Ad-PD patients of the 89 Ad-PD index patients (allele frequency, 1.7%) and three of the 73 sPD patients (allele frequency, 2.2%). The previously reported nonsynonymous variant p.G2385R was identified in 2 of the 11 Japanese Ad-PD families (allele frequency, 9%), but not found in the 19 Caucasian families. Because recent studies in Taiwan have shown a higher frequency of p.G2385R in sPD than in controls (5.0 vs 2.5%, P=0.012),16 the frequencies of p.G2385R in controls, sPD and Ad-PD were further analyzed as described below.

(a) Scan images of heterozygous LRRK2 p.N1221K putative pathogenic mutation. Each column shows a base position, and each row shows a base call. In the center position, the base call was C in the control, whereas in the patients, the base calls were heterozygous C and G (upper panel). The mutation was confirmed by direct nucleotide sequencing. (b) Conservation of LRRK2 amino-acid sequences among different animal species. The arginine residue at codon 1221 is highly conserved among species (shown in red). A full color version of this figure is available at the Journal of Human Genetics journal online.
Among the variants identified in Caucasian families, two (p.I1371V and p.G2019S) were initially missed in the analysis using GDAS software and subsequent visual inspection of the signals of undetermined base calls, but were independently identified by direct nucleotide sequence analyses. Retrospective analysis of the microarray signals of these two variants showed that the signals for p.I1371V could have been detected as a heterozygous variant, whereas the signals for p.G2019S would have been difficult to determine considering that one of the three patients with p.G2019S was called as having a homozygous wild-type sequence using GDAS software.
Association of p.G2385R with Ad-PD
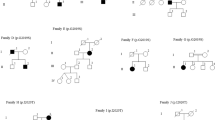
As mentioned above, we identified p.G2385R in two Japanese Ad-PD families. We first analyzed the frequency of p.G2385R in another data set of 89 Japanese Ad-PD families independent of the 11 Ad-PD families. We found that the seven index patients from the 89 Ad-PD families had p.G2385R. Among the seven Ad-PD families with p.G2385R, DNA samples were available from other pedigree members including affected patients in two families (families 2938 and 3045). In the two families (families 2955 and 2932), DNA samples from only other unaffected family members were available (Figure 2). p.G2385R did not show complete cosegregation with PD. Although cosegregation was suggested in family 3045, one affected patient did not carry p.G2385R in the family 2938. Furthermore, unaffected parents of the two other families (families 2955 and 2932) also carried the p.G2385R variant in a heterozygous state. Nevertheless, the frequency in 18 index patients of the 100 Ad-PD families of p.G2385R was significantly higher than that in controls (allele frequency, 9.0 vs 2.1%) (χ2=16.32, P=5.34 × 10−5) (Table 3). p.G2385R has recently been reported to be a polymorphism present in the Japanese control population, but overrepresented in sPD patients compared with controls (6.0 vs 2.4%, P=1.24 × 10−4).17 Given this report, we then analyzed the frequencies of p.G2385R in 73 sPD patients and 233 controls (Table 3). The frequency of the p.G2385R variant in sPD also tended to be higher than that in controls (allele frequency, 3.4 vs 2.1%), although the difference did not reach statistical significance (χ2=0.76, P=0.38) (Table 3). The mean age at onset of Ad-PD in patients carrying p.G2385R was 55.3±6.1 (18–67), which was not significantly different from that in patients not carrying the variant (51.8±13.0, P=0.49). The mean age at onset of sPD in patients carrying the p.G2385R variant was 60.2±6.9 (52–71) years, which was similarly not significantly different from that of sPD in patients not carrying the variant (51.3±15.0, P=0.19). The two Ad-PD and five sPD patients with p.G2385R whose detailed clinical information was available showed a good response to levodopa therapy and no atypical features.

Pedigree charts in which cosegregation of LRRK2 p.G2385R was examined. Affected individuals are indicated with black symbols. The proband is indicated with an arrow. Unaffected individuals are indicated with open symbols. Slashed symbols indicate deceased subjects. Genotype of each member available for cosegregation analysis is shown at each position. G: wild-type allele, A: p.G2385R allele (rs34778348:G>A).
Discussion
This study demonstrated that the spectrum and frequency of LRRK2 variants vary with ethnic background. In Caucasians, p.G2019S is the most frequent in both Ad-PD and sPD, accounting for ∼5% of Ad-PD patients and 1.6% of sPD patients.8, 9, 18 Many of the patients from European populations share a common haplotype, suggesting that they have a common founder originating in the Near East at least 4000 years ago.19, 20 In contrast, no single mutation has been reported as being predominant in Asian populations. Although three Japanese index patients with p.G2019S were reported,21, 22 we did not detect p.G2019S in our Japanese Ad-PD cohort, consistent with previous reports, showing that p.G2019S is rare in Asian cohorts.10, 11, 18, 23 Indeed, the substantial differences in allele frequencies of the synonymous LRRK2 polymorphisms (rs10878245, rs1427263, rs11176013, rs17466213 and rs3761863) highlight the genetic diversity between the Japanese and Caucasian Ad-PD patients (Table 2). These findings raise the possibility that mutational analysis focusing on previously identified mutation hotspots may underestimate the incidence of mutations; therefore, a comprehensive analysis of every coding exon is warranted.
Our comprehensive analysis of LRRK2 revealed 22 variants in 30 Ad-PD families, supporting the usefulness of the DNA microarray-based resequencing system. However, it should also be noted that data analysis using GDAS software and subsequent visual inspection of the signals of undetermined base calls may miss variants, as experienced in the cases of p.I1371V and p.G2019S. Improvement of software and careful inspection of signals are important for accurate mutational detection.
In the Japanese Ad-PD families, we identified a novel nonsynonymous variation, p.N1221K. Although cosegregation could not be confirmed, it is possibly a pathogenic mutation for the following reasons: (1) p.N1221K was not found in 466 control chromosomes, (2) the amino-acid residue N1221 was highly conserved among different animal species and (3) it is located in the LRR domain, where some likely pathogenic mutations are clustered.24, 25, 26 The LRR domain resides in many proteins with diverse functions and provides a structural framework for the formation of protein–protein interactions.27 The substitution of a charged amino-acid lysine for a neutral asparagine may disrupt protein–protein interactions, resulting in the dysfunction of the LRRK2 protein. Identification of additional pedigrees harboring the mutation, cosegregation analysis and functional studies would be necessary to confirm the pathogenicity of the mutation.
The age at onset of the patient with p.N1221K was earlier than those of patients with previously reported mutations of LRRK2 (the mean age at onset was in the range of 57–59 years).7, 9, 25 Considering that p.G2019S is the most frequent among the known mutations, the overall clinical presentation of patients with LRRK2 mutations may largely reflect that of patients with p.G2019S. Interestingly, a previously reported case of a patient with the p.S1228T mutation, located in the same exon and domain as in p.N1221K, showed a similarly early age at onset to the patient with p.N1221K, who developed symptoms at the age of 49.25 It will be important to accumulate further detailed clinical information on patients with LRRK2 mutations to evaluate the genotype–phenotype correlations.
In the Caucasian Ad-PD families, we identified two nonsynonymous variants, p. I1371V and p. R1514Q, both of which were initially reported as a putative pathogenic mutation.28, 29 However, subsequent studies have revealed that these variants were identified in controls or not cosegregated within Ad-PD families, implying that they are not associated with an increased risk of the disease.30, 31, 32 It is often difficult to interpret the pathogenicity of each rare nonsynonymous variant in LRRK2 because this gene is highly polymorphic and numerous nonsynonymous variants have been identified with diverse frequencies among different ethnic backgrounds. In this study, we could not conclude that the two nonsynonymous variants were pathogenic because large families appropriate for cosegregation analysis were not obtained and control data were not available, and further studies are required to evaluate the relevance of the variants to the pathogenesis of PD.
Of note, a known nonsynonymous variant R50H was found homozygously in all the pedigrees with the allele frequency of 100% in this study. Although the variant was initially registered as a nonsynonymous single-nucleotide polymorphism, a subsequent study on the comprehensive sequencing of LRRK2 concluded that this variant should be a mistake in the consensus human genome sequence because it was present in all the cases and controls in their study.33 The present study further supports the above conclusion that the consensus amino acid at position 50 in LRRK2 protein is histidine instead of arginine. Future studies on the mutational analysis for LRRK2 should take these results into account.
Heterozygosity for the p.G2385R variant has recently been demonstrated to be significantly more frequent in PD patients than in controls in Asian populations.16, 17, 34, 35, 36, 37, 38, 39 One of these studies included 26 patients with a familial history of PD, among which six patients were p.G2385R carriers.40 A meta-analysis on the basis of these studies involving 2205 PD patients and 1817 controls demonstrated average carrier rates of 9.3% in PD and 4% in controls.41 In this study, the frequency of the p.G2385R variant did not reach statistical significance between sPD patients and controls, probably due to relatively small sample size.
Intriguingly, we observed a significant overrepresentation of the p.G2385R variant in Ad-PD index patients compared with that in controls, although no complete cosegregation was observed. Note that the frequency of the p.G2385R variant in our control cohort was the same as that in a previous report (2.4%).17 In addition, the frequency of the p.G2385R variant also tended to be higher in Ad-PD patients than in sPD patients in accordance with a previous study.40 Taken together, this study reinforced the notion that p.G2385R is a potential risk factor for PD. However, lack of evidences on the cosegregation suggested that other genetic factors should also be involved in the disease pathogenesis in the pedigrees with p.G2385R mutation. Analyses of p.G2385R in combination with recently identified genetic risk factors for PD such as SNCA42, 43, 44 and GBA45, 46 would be important for evaluating the combined effects of these factors as well as for elucidating the molecular pathophysiology of PD.
References
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 (1997).
Kitadam, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 (1998).
Leroy, E., Boyer, R., Auburger, G., Leube, B., Ulm, G., Mezey, E. et al. The ubiquitin pathway in Parkinson's disease. Nature 395, 451–452 (1998).
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259 (2003).
Paisán-Ruíz, C., Jain, S., Evans, E. W., Gilks, W. P., Sim, J., van der Brug, M. et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44, 595–600 (2004).
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S. et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 (2004).
Healy, D. G., Falchi, M., O’Sullivan, S. S., Bonifati, V., Durr, A., Bressman, S. et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. 7, 583–590 (2008).
Di Fonzo, A., Rohe, C. F., Ferreira, J., Chien, H. F., Vacca, L., Stocchi, F. et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet 365, 412–415 (2005).
Gilks, W. P., Abou-Sleiman, P. M., Gandhi, S., Jain, S., Singleton, A., Lees, A. J. et al. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet 365, 415–416 (2005).
Lu, C. S., Simons, E. J., Wu-Chou, Y. H., Fonzo, A. D., Chang, H. C., Chen, R. S. et al. The LRRK2 I2012T, G2019S, and I2020T mutations are rare in Taiwanese patients with sporadic Parkinson's disease. Parkinsonism Relat. Disord. 11, 521–522 (2005).
Tan, E. K., Shen, H., Tan, L. C., Farrer, M., Yew, K., Chua, E. et al. The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson's disease patients. Neurosci. Lett. 384, 327–329 (2005).
Kay, D. M., Zabetian, C. P., Factor, S. A., Nutt, J. G., Samii, A., Griffith, A. et al. Parkinson's disease and LRRK2: frequency of a common mutation in U.S. movement disorder clinics. Mov. Disord. 21, 519–523 (2006).
Paisan-Ruiz, C., Lang, A. E., Kawarai, T., Sato, C., Salehi-Rad, S., Fisman, G. K. et al. LRRK2 gene in Parkinson disease: mutation analysis and case control association study. Neurology 65, 696–700 (2005).
Cutler, D. J., Zwick, M. E., Carrasquillo, M. M., Yohn, C. T., Tobin, K. P., Kashuk, C. et al. High-throughput variation detection and genotyping using microarrays. Genome Res. 11, 1913–1925 (2001).
Warrington, J. A., Shah, N. A., Chen, X., Janis, M., Liu, C., Kondapalli, S. et al. New developments in high-throughput resequencing and variation detection using high density microarrays. Hum. Mutat. 19, 402–409 (2002).
Di Fonzo, A., Wu-Chou, Y. H., Lu, C. S., van Doeselaar, M., Simons, E. J., Rohe, C. F. et al. A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson's disease risk in Taiwan. Neurogenetics 7, 133–138 (2006).
Funayama, M., Li, Y., Tomiyama, H., Yoshino, H., Imamichi, Y., Yamamoto, M. et al. Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport 18, 273–275 (2007).
Funayama, M., Hasegawa, K., Ohta, E., Kawashima, N., Komiyama, M., Kowa, H. et al. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann. Neurol. 57, 918–921 (2005).
Lesage, S., Patin, E., Condroyer, C., Leutenegger, A. L., Lohmann, E., Giladi, N. et al. Parkinson's disease-related LRRK2 G2019S mutation results from independent mutational events in humans. Hum. Mol. Genet. 19, 1998–2004 (2010).
Zabetian, C. P., Hutter, C. M., Yearout, D., Lopez, A. N., Factor, S. A., Griffith, A. et al. LRRK2 G2019S in families with Parkinson disease who originated from Europe and the Middle East: evidence of two distinct founding events beginning two millennia ago. Am. J. Hum. Genet. 79, 752–758 (2006).
Tomiyama, H., Li, Y., Funayama, M., Hasegawa, K., Yoshino, H., Kubo, S. et al. Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson's disease patients from 18 countries. Mov. Disord. 21, 1102–1108 (2006).
Zabetian, C. P., Morino, H., Ujike, H., Yamamoto, M., Oda, M., Maruyama, H. et al. Identification and haplotype analysis of LRRK2 G2019S in Japanese patients with Parkinson disease. Neurology 67, 697–699 (2006).
Cho, J. W., Kim, S. Y., Park, S. S., Kim, H. J., Ahn, T. B., Kim, J. M. et al. The G2019S LRRK2 mutation is rare in Korean patients with Parkinson's disease. Can. J. Neurol. Sci. 34, 53–55 (2007).
Paisán-Ruíz, C., Nath, P., Washecka, N., Gibbs, J. R. & Singleton, A. B. Comprehensive analysis of LRRK2 in publicly available Parkinson's disease cases and neurologically normal controls. Hum. Mutat. 29, 485–490 (2008).
Berg, D., Schweitzer, K., Leitner, P., Zimprich, A., Lichtner, P., Belcredi, P. et al. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson's disease. Brain 128, 3000–3011 (2005).
Skipper, L., Shen, H., Chua, E., Bonnard, C., Kolatkar, P., Tan, L. C. et al. Analysis of LRRK2 functional domains in nondominant Parkinson disease. Neurology 65, 1319–1321 (2005).
Kobe, B. & Kajava, A. V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 11, 725–732 (2001).
Mata, I., Kachergus, J., Taylor, J., Lincoln, S., Aasly, J., Lynch, T. et al. Lrrk2 pathogenic substitutions in Parkinson's disease. Neurogenetics 6, 171–177 (2005).
Lesage, S., Condroyer, C., Lannuzel, A., Lohmann, E., Troiano, A., Tison, F. et al. Molecular analyses of the LRRK2 gene in European and North African autosomal dominant Parkinson's disease. J. Med. Genetics 46, 458–464 (2009).
Toft, M., Mata, I., Ross, O. A., Kachergus, J., Hulihan, M. M., Haugarvoll, K. et al. Pathogenicity of the Lrrk2 R1514Q substitution in Parkinson's disease. Mov. Disord. 22, 389–392 (2007).
Nichols, W. C., Marek, D. K., Pauciulo, M. W., Pankratz, N., Halter, C. A., Rudolph, A. et al. R1514Q substitution in Lrrk2 is not a pathogenic Parkinson's disease mutation. Mov. Disord. 22, 254–256 (2007).
Jasinska-Myga, B., Kachergus, J., Vilarino-Guell, C., Wider, C., Soto-Ortolaza, A. I., Kefi, M. et al. Comprehensive sequencing of the LRRK2 gene in patients with familial Parkinson's disease from North Africa. Mov. Disord. 25, 2052–2058 (2010).
Paisán-Ruiz, C. LRRK2 gene variation and its contribution to Parkinson disease. Hum. Mutat. 30, 1153–1160 (2009).
Tan, E. K., Zhao, Y., Skipper, L., Tan, M. G., Di Fonzo, A., Sun, L. et al. The LRRK2 Gly2385Arg variant is associated with Parkinson's disease: genetic and functional evidence. Hum. Genet. 120, 857–863 (2007).
Kim, J. M., Lee, J. Y., Kim, H. J., Kim, J. S., Shin, E. S., Cho, J. H. et al. The LRRK2 G2385R variant is a risk factor for sporadic Parkinson's disease in the Korean population. Parkinsonism Relat. Disord. 16, 85–88.
Tan, E. K., Peng, R., Teo, Y. Y., Tan, L. C., Angeles, D., Ho, P. et al. Multiple LRRK2 variants modulate risk of Parkinson disease: a Chinese multicenter study. Hum. Mutat. 31, 561–568 (2010).
An, X. K., Peng, R., Li, T., Burgunder, J. M., Wu, Y., Chen, W. J. et al. LRRK2 Gly2385Arg variant is a risk factor of Parkinson's disease among Han-Chinese from mainland China. Eur. J. Neurol. 15, 301–305 (2008).
Chan, D. K., Ng, P. W., Mok, V., Yeung, J., Fang, Z. M., Clarke, R. et al. LRRK2 Gly2385Arg mutation and clinical features in a Chinese population with early-onset Parkinson's disease compared to late-onset patients. J. Neural. Transm. 115, 1275–1277 (2008).
Zabetian, C. P., Yamamoto, M., Lopez, A. N., Ujike, H., Mata, I. F., Izumi, Y. et al. LRRK2 mutations and risk variants in Japanese patients with Parkinson's disease. Mov. Disord. 24, 1034–1041 (2009).
Farrer, M. J., Stone, J. T., Lin, C. H., Dachsel, J. C., Hulihan, M. M., Haugarvoll, K. et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson's disease in Asia. Parkinsonism Relat. Disord. 13, 89–92 (2007).
Tan, E. K. The role of common genetic risk variants in Parkinson disease. Clin. Genet. 72, 387–393 (2007).
Satake, W., Nakabayashi, Y., Mizuta, I., Hirota, Y., Ito, C., Kubo, M. et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 41, 1303–1307 (2009).
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D. et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 41, 1308–1312 (2009).
Mizuta, I., Satake, W., Nakabayashi, Y., Ito, C., Suzuki, S., Momose, Y. et al. Multiple candidate gene analysis identifies alpha-synuclein as a susceptibility gene for sporadic Parkinson's disease. Hum. Mol. Genet. 15, 1151–1158 (2006).
Mitsui, J., Mizuta, I., Toyoda, A., Ashida, R., Takahashi, Y., Goto, J. et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch. Neurol. 66, 571–576 (2009).
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N. Engl. J. Med. 361, 1651–1661 (2009).
Acknowledgements
The Canadian portion of this study was supported in part by a Center of Excellence Grant from the National Parkinson Foundation (AL, CM) and Canadian Institutes of Health Research (PH, ER). This work was also supported by the High-Tech Research Center Project, a Grant-in-Aid for Scientific Research (to NH, 17390256, and to HT, 21591098), and a Grant-in-Aid for Scientific Research on Priority Areas (to NH, 08071510) from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Seki, N., Takahashi, Y., Tomiyama, H. et al. Comprehensive mutational analysis of LRRK2 reveals variants supporting association with autosomal dominant Parkinson's disease. J Hum Genet 56, 671–675 (2011). https://doi.org/10.1038/jhg.2011.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.79
Keywords
This article is cited by
-
Molecular epidemiology and clinical spectrum of hereditary spastic paraplegia in the Japanese population based on comprehensive mutational analyses
Journal of Human Genetics (2014)
-
The screening of the 3′UTR sequence of LRRK2 identified an association between the rs66737902 polymorphism and Parkinson’s disease
Journal of Human Genetics (2014)
