
Abstract
There are 68 sex-linked syndromes that include hearing loss as one feature and five sex-linked nonsyndromic deafness loci listed in the OMIM database. The possibility of additional such sex-linked loci was explored by ascertaining three unrelated Pakistani families (PKDF536, PKDF1132 and PKDF740) segregating X-linked recessive deafness. Sequence analysis of POU3F4 (DFN3) in affected members of families PKDF536 and PKDF1132 revealed two novel nonsense mutations, p.Q136X and p.W114X, respectively. Family PKDF740 is segregating congenital blindness, mild-to-profound progressive hearing loss that is characteristic of Norrie disease (MIM#310600). Sequence analysis of NDP among affected members of this family revealed a novel single nucleotide deletion c.49delG causing a frameshift and premature truncation (p.V17fsX1) of the encoded protein. These mutations were not found in 150 normal DNA samples. Identification of pathogenic alleles causing X-linked recessive deafness will improve molecular diagnosis, genetic counseling and molecular epidemiology of hearing loss among Pakistanis.
Similar content being viewed by others


Introduction
X-linked nonsyndromic hearing impairment is genetically heterogeneous with a prevalence of 1–5% in different populations.1, 2 To date, five loci for nonsyndromic hearing loss (DFN2, DFN3, DFN4, DFN6 and AUNX) have been reported and genes for two of them, PRPS1 (DFN2) and POU3F4 (DFN3), have been identified.3, 4 PRPS1 encodes phospho-ribosylpyrophosphate synthetase 1 (PRS-I; EC 2.7.6.1), which is essential for the first step of nucleotide synthesis pathway. Missense mutations that dysregulate PRS-I enzymatic activity are also associated with X-linked Charcot–Marie–Tooth disease-5 (MIM 311070), Arts syndrome (MIM 301835) and PRS-I superactivity disorder (MIM 300661).4 POU3F4 (DFN3) mutations are the most common cause of sex-linked nonsyndromic deafness.2, 3 POU3F4 encodes a transcription factor, which belongs to the POU-domain family and is comprised of two subdomains, POU-specific domain and a homeodomain.5 To date, 30 mutations, including two nonsense alleles, in the coding region of the POU3F4 have been identified as a cause of nonsyndromic hearing loss in DFN3 families.6
X-linked hearing loss can also manifest as part of a syndrome. For example, Norrie disease (ND) is characterized by congenital or early childhood blindness and progressive mental retardation with reduced penetrance.7, 8 Progressive sensorineural hearing loss has been reported in about one-third of the ND cases.7, 8 Mutations of NDP-encoding norrin, a protein of 133 residues, were identified in subjects with ND.8, 9, 10 A total of 55 different pathogenic alleles, including missense, nonsense, splice site, insertion and deletions, have been identified in ND subjects of North American, European and Japanese ancestries (http://www.retina-international.com/sci-news/ndgmut.htm).
To understand the genetic causes of X-linked hearing impairment in the Pakistani population, we ascertained families segregating sex-linked hearing loss in this population. Here, we report three large consanguineous Pakistan families, PKDF536, PKDF1132 and PKDF740, segregating X-linked recessive hearing loss. Clinical investigation revealed nonsyndromic deafness segregating in families PKDF536 and PKDF1132. We found two novel nonsense mutations, p.Q136X and p.W114X, of POU3F4 co-segregating with hearing loss in these two families, respectively. Affected individuals of family PKDF740 were diagnosed with ND and mutational analysis of NDP revealed a one base pair deletion, c.49delG, that results in a translation frameshift and a predicted premature truncation of 117 residues of norrin.
Materials and methods
Family enrollment and diagnosis
This study was approved by the Institutional Review Board (IRB) at the National Centre of Excellence in Molecular Biology (NCEMB), Lahore, Pakistan (FWA00001758), the Combined Neuroscience IRB at the National Institutes of Health, USA (OH-93-N-016) and, the IRB Committee at the Cincinnati Children's Hospital Research Foundation, USA (2009-0684). Written informed consent was obtained from all participating individuals and parents of minor subjects. The pedigree structure and information about consanguinity is based on interviews with family members.
Clinical evaluation
Participating subjects were evaluated by medical history interviews. A physical examination was performed on at least one hearing-impaired individual from each family. The degree of hearing loss was assessed by pure tone audiometry using air conduction at frequencies ranging from 250–8000 Hz. Computational tomography scan was performed on members of families PKDF536 and PKDF1132 to access POU3F4 mutation-linked middle-ear anomalies. Detailed funduscopic examination of some affected individuals ruled out obvious signs of retinopathy. Magnetic resonance imaging and brightness scan was performed to characterize the ocular phenotype of ND in PKDF741. During magnetic resonance imaging, multi-planer imaging was performed through both orbits acquiring T1/T2-weighted and proton density sequences. Vestibular function was evaluated by tandem gait and Romberg testing.
DNA isolation, linkage analysis and sequencing
Venous blood samples were obtained and genomic DNA was extracted using a standard protocol. Short tandem repeat (ABI Prism Linkage Mapping Set v2.5, Applied Biosystems Foster City, CA, USA) markers spaced at ∼10 cM and also covering the X-chromosome were used for linkage analysis. Markers were amplified on a Gene Amp PCR system 2700, analyzed on ABI Prism 3730 Genetic Analyzer and the alleles were assigned using GeneMapper software (Applied Biosystems). Sequencing primers of POU3F4 and NDP were designed from the flanking region of each exon using Primer3 web utility (http://frodo.wi.mit.edu/primer3/). Sequencing reactions and mutational analyses were performed as described previously.11
Results
Family PKDF536
The proband V:1 was first enrolled through a school for hearing impaired individuals. A family history revealed the presence of additional affected members all of whom were male (Figure 1a). According to the parents or other family members, the time of hearing loss was pre-lingual. Air and bone conduction audiometery of two affected individuals of this family showed severe-to-profound mixed hearing loss (Figure 1c).

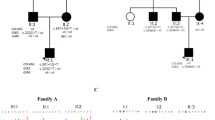
Family PKDF536. (a) Pedigree of PKDF536 with seven affected individuals in four sibships. Circles indicate females and fully shaded squares represent males with hearing loss; and the central dot in the female symbol indicate the carrier of the affected X-chromosome. (b, c) Representative pure-tone air and bone conduction audiometric data of affected individual IV:11 (24 years old) along with a normal-hearing subject, III:7 (60 years old). Bilateral severe-to-profound mixed hearing loss with a significant low-frequency conductive hearing loss was observed in the affected individuals of family PKDF536. (d) Sequence chromatogram of pathogenic variant, c.406C>T (p.Q136X), along with a homozygous wild-type allele.
X-chromosome haplotype analysis revealed co-segregation of hearing loss in family PKDF536 with DFN3-linked short tandem repeat markers (data not shown). DFN3 is caused by mutations in POU3F4 gene as well as by inversions, duplications and deletions upstream of this gene.2, 3, 12, 13, 14, 15, 16, 17 Sequence analysis of POU3F4 in affected males of family PKDF536 revealed a novel transversion mutation (c.406C>T), which results in a nonsense mutation (p.Q136X; Figure 1d). The p.Q136X mutation is predicted to truncate the protein before the POU-specific and homeobox domains. All affected males had the c.406C>T mutation, whereas the female carrier had normal hearing (Figure 1b). c.406C>T was absent in 150 DNA samples from normal-hearing Pakistani individuals and was also absent from the 1000 Genomes database of human genetic variation.
High-resolution temporal bone computational tomography scan of affected individual IV:11 of PKDF536 showed bilaterally minimal widening of the lateral ends of the internal auditory canals (Figure 2). The basal and middle turns of cochlea, semicircular canals, vestibules and ossicules were normally developed. But the mid and apical turns are not clearly partitioned in individual IV:11 (Figure 2c). A wide modiolus at the junction of the cochlea with the internal auditory canal was also observed (Figure 2e).

Representative axial and bone-window images from computed tomographic (CT) scans from individual IV:11 of family PKDF536 with p.Q136X mutation in POU3F4 along with a control subject (with normal hearing, normal CT anatomy and no known deafness-causing genetic mutation). (a–d) Highlights the subtle cochlear hypoplasia associated with the POU3F4 genotype in comparison to the control subject. Although a clear basal turn of the cochlea is apparent in both affected and control subjects (coch: cochlea and arrows in a–d), the mid and apical turns are not clearly partitioned in the affected individual. (e, g) Illustrate the modiolar and internal auditory canal anomalies typical in individuals with the POU3F4 mutation. (f, h) Normal morphology is seen in the control subject. (e) The extremely wide modiolus at the junction of the cochlea with the internal auditory canal (contrast with the typical modiolus and internal auditory canal shown in f). (h) CT scans from a normal-hearing individual to provide a comparison of the dysmorphic internal auditory canals seen in the affected individual (g). This combination of a dilated and dysmorphic internal auditory canal along with the very broad modiolus have been postulated to be the cause of a ‘stapes gusher’ seen at the time of surgery in patients with POU3F4 mutations.
Family PKDF1132
A multi-generation family (Figure 3a) segregating pre-lingual hearing loss was ascertained from Punjab province of the Pakistan. Pure tone audiometry of the three participating hearing-impaired individuals showed bilateral severe-to-profound mixed hearing loss (Figure 3c). A high-resolution temporal bone computational tomography scan was performed on individual VI:4 of family PKDF1132 (Figure 3a). The internal auditory canal had an unusual bulbous morphology and it directs anteromedially rather than continuing in a straight direction. Bony deficiency at the base of cochlea was also observed (Figure 3d).

Family PKDF1132. (a) Pedigree of family PKDF1132 with six affected individuals present in three sibships. Circles indicate females and fully shaded squares represent males with hearing loss; and the central dot in a female symbol indicates a carrier of a X-linked mutation. (b, c) Representative pure-tone air and bone conduction audiometric data of affected individual IV:4 (16 years old male) along with a normal-hearing subject, III:2 (49 years old female). Severe-to-profound mixed hearing loss in both ears was observed in the affected individuals of family PKDF536. (d) Temporal bone computed tomography of individual IV:4. Basal turn of the cochlea was incompletely separated from the internal auditory canal (IAC), and seemed to be a continuation of the IAC. Typically, a bulbous dilation of the lateral end of the IAC (arrow) was identified. (e) Sequence chromatogram of pathogenic variant, c.341G>A (p.W114X), along with a homozygous wild-type allele.
Linkage analysis studies revealed common alleles among all of the affected individuals for DFN3-linked short tandem repeat markers (data not shown). Sequence analysis of POU3F4 in affected members of family PKDF1132 revealed a novel transition mutation (c.341G>A), which results in a nonsense mutation (p.W114X; Figure 3e). The c.341G>A transition mutation was absent in 150 DNA samples from normal-hearing Pakistani individuals and in the 1000 Genomes database of human genetic variation. The truncated protein due to p.W114X is predicted to lack all of the known domains of POU3F4.
Family PKDF740
Family PKDF740 has four affected individuals in two non-consanguineous sibships (Figure 4a). All of the affected individuals had congenital, bilateral blindness and progressive mild-to-profound hearing loss (Figures 4c–e) as well as microphthalmia, clinical features consistent with ND.7 Brightness scan of individual III:1 of family PKDF740 revealed severe microphthalmia with retinal dysplasia and disorganization of intraocular contents in the right eye. A focal area of calcification posteriorly may represent a pseudoglioma. The scan of the left eye showed severe microphthalmia with three foci of increased sonoreflectivity corresponding to a dense cataract and total funnel retinal detachment with calcific shadowing blocking the contour of the posterior curvature of the globe. Magnetic resonance imaging of individual III:1 showed that anteroposterior dimensions of the globes are microophthalmic and were 0.5 cm at right and 0.8 cm (normal range: 2.38–3.11 cm) on the left side with hypointense masses projecting anteriorly from the optic nerves (Figure 5). These represent funnel retinal detachment and/or pseudogliomas (dysplastic retina/optic nerve mass). No evidence of conal or extra-conal mass. Lacrimal glands and optic nerves are normal in appearance. Brain parenchyma was normal, but the optic nerves, chiasm and tracts were small, if present at all, indicating either primary hypoplasia or secondary postaxonal degeneration (Figure 5). Four of the five available females have normal hearing and vision. One female (III:2) has normal hearing (Figure 4b) but brightness scan images of her right eye showed severe microphthalmia, phthisis and discoid calcific lens with absent anterior chamber. Strong acoustic shadowing prevents evaluation of the remaining posterior segment. Brightness scan of the left eye revealed severe microphthalmia and phthisis. Ocular contents are not discernible posterior to calcification.

Family PKDF740. (a) Pedigree of family PKDF740 with four affected individuals present in two sibships. Circles indicate females and fully shaded squares represent males with hearing loss; and the central dot in a female symbol indicates a carrier of a X-linked mutation. (b, c) Pure-tone air conduction audiometric data of three affected individuals along with a normal-hearing subject, III:2 (19 years old female). (b) Normal-hearing thresholds were observed across all frequencies for individual III:2. (c) Mild-to-profound hearing loss was observed in individual II:3. (d) Severe hearing impairment across all frequencies were seen in both ears of individual III:1. (e) Oldest affected individual, II:5, of family PKDF740 has severe to profound hearing loss in both ears. (f) Sequence chromatogram of pathogenic variant, c.49delG (p.V17fsX1), along with a homozygous wild-type allele.

Magnetic resonance imaging imaging of individual III:1 of family PKDF740. (a) Axial T2 image of microphthalmic phthisical globes with atrophic optic nerves exiting the globes posteriorly bilaterally. (b–d) Optic tracts and chiasm were not discernable on axial T2 (b), coronal T1 (c), and coronal T2 (d) images, indicating either primary hypoplasia or postaxonal degeneration.
ND appears to be genetically homogenous as it is caused by mutations in NDP.8, 9, 10 Mutational analysis of all the exons of NDP in the DNA samples from affected individuals of family PKDF740 revealed a novel frameshift mutation c.49delG in exon 2. Deletion of a single nucleotide is predicted to cause a translation-reading frameshift and premature truncation of the encoded protein, p.V17fsX1. This mutation was not found in 150 DNA samples from normal-hearing Pakistani individuals and in the 1000 Genomes database of human genetic variation.
Discussion
DFN3 (POU3F4)
Five DFN loci have been mapped and genes for two of them, PRPS1 (DFN2) and POU3F4 (DFN3), have been identified.3, 4 Among the known loci, DFN3 accounts for ∼50% of nonsyndromic X-linked hearing loss.2 Clinically, DFN3 is characterized by congenital sensorineural or mixed hearing loss and perilymphatic gusher occurring during stapes surgery.18 POU3F4, mutated in DFN3 families, encodes a polypeptide of 361 amino acids with bi-partite DNA-binding domains (POU and homeobox), and thus belongs to the POU family of transcription factors.5, 8, 9, 10 A total of 30 different mutations, including nonsense mutations, deletions, inversions and duplication, within or upstream of the POU3F4 gene have been reported.6 Most of these alleles have been identified in the Caucasian populations and very few studies have described in the Asian population.14, 19 In this study, we identified two novel nonsense mutations (p.Q136X) and (p.W114X) in POU3F4 as a cause of nonsyndromic recessive hearing loss in two large Pakistani families. Both of these mutations (p.Q136X and p.W114X) are predicted to cause premature truncation of the POU3F4 protein prior to the POU and homeobox domains, hence likely representing functional null alleles. Some studies have reported the hearing loss in female siblings or mothers of affected males with mutations in POU3F4.20 However, in our two families we did not observe hearing loss in heterozygous mothers (Figures 1 and 3). Air and bone conduction audiometry of both families revealed the severe-to-profound bilateral mixed hearing loss in all the affected individuals of both families along with structural abnormalities of the cochlea, a phenotype consistent with previous studies.6, 12, 13, 16, 21 Deficiency of POU3F4-associated defects of the bony labyrinth includes dilation of the internal auditory canal, partial hypoplasia of the spiral limbus, scala tympani and strial fibrocytes, with incomplete penetrance of a reduction in the cochlear turns. Outward pressure of perilymphatic fluid on the oval window coupled with defects in the size and shape of stapes footplate further compromise ossicular movement and collectively, these anomalies result in a conductive hearing loss in DFN3 patients.21, 22
The management of patients with mixed hearing loss depends on the degree of the overall hearing loss, as well as the relative contributions of the sensorineural and conductive hearing loss components. For those patients with milder hearing impairments (whether conductive or sensorineural), or if the hearing loss is predominantly sensorineural, then hearing aids are often a first line recommendation. However, if the overall hearing impairment is more severe, and there is a substantial conductive component, then consideration is given to a surgical intervention to address the conductive component. A pertinent cautionary aspect in this line of management is the possibility of a ‘stapes gusher’ that is well described in some patients.6, 12, 13 In this phenomenon, surgical removal or manipulation of the stapes can result in a robust flow of perilymph from the oval window. Such brisk outflow of cochlear fluids can result in an immediate deafness along with concomitant complaints of vertigo and tinnitus. Recently, some patients with bilateral mixed hearing losses but serviceable bone conduction thresholds have been shown to benefit from bone anchored hearing aid technologies. Such interventions are attractive in avoiding surgical manipulation of the stapes and thus avoiding the risk of a gusher, but still providing effective gain via bone conduction technologies.
Norrie disease
ND is a severe X-linked recessive disorder with clinical manifestations of congenital or very early childhood blindness due to degenerative and proliferative changes of the neuroretina. About 50% of ND patients show progressive mental disorder, often with psychotic features, and approximately one-third of the cases develop bilateral, progressive, sensorineural deafness in the second decade.7 In addition, some patients have more complex phenotypes, including growth failure, seizures, profound psychomotor retardation, delayed sexual maturation, stereotypies, microcephaly, cardiovascular abnormalities and idiopathic pulmonary arterial hypertension, and are thus diagnosed with ‘Norrie-plus’ syndrome.8, 9, 10, 23, 24, 25, 26, 27, 28, 29, 30, 31 The Norrie-plus syndrome is associated with microdeletions encompassing NDP and neighboring genes at Xp11.3-11.4, including MAO-A, MAO-B and EFHC2.23 The NDP gene encodes norrin, a secreted cysteine-rich protein of 133 residues that belongs to the cystine knot growth factor family.5, 9 A total of 55 different pathogenic alleles of NDP have been reported, including the 13 private mutations found in the Asian populations, mainly Japanese and Chinese families. However, to our knowledge, no NDP mutation in the Indo–Pak subcontinent population has been reported. Sequence analysis of NDP in DNA samples from affected individuals of Pakistani family PKDF740 revealed a novel single-base pair deletion, c.49delG, in exon 2, which is predicted to cause premature truncation (p.V17fsX1) of Norrin protein, and thus is likely to be a functional null allele.
On the basis of the mutational screening of 109 patients with pediatric vitreoretinopathies, Wu et al.32 defined a genotype–phenotype correlation, in which the NDP mutations disrupt the cysteine-knot motif resulting in severe retinal dysgenesis and a diagnosis of ND, whereas patients with noncysteine mutations have varying degrees of avascular peripheral retina, extraretinal vasculature and subretinal exudate. In family PKDF740, congenital blindness due to microphthalmia co-segregated with mild-to-profound, progressive hearing loss. One obligatory carrier female III:2 (30 years) share the ocular symptoms with affected individuals but had normal air and bone conduction thresholds (Figure 4b). The incomplete penetrance of the clinical symptoms in the obligate carrier females has been documented in a few studies.33, 34, 35 This unusual phenotype in a female has been attributed to Lyonization or an adverse effect of the mutant copy of the protein on the retina during development.33, 34, 35 In family PKDF740, there were no obvious signs of mental retardation, although this phenotype was not methodically pursued at the time family members were ascertained.
Incomplete retinal vascularization, retinal folding and detachment, microphthalmia, alteration in the composition of vitreous body and vitreoretinal hemorrhages, the highly penetrant clinical symptoms of ND, bear significant phenotypic overlap with familial exudative vitreoretinopathy (FEVR). FEVR is a genetically heterogeneous disease and can manifest under a variety of inheritance models.10, 36, 37 Mutations in FDZ4 and LRP5 that encode frizzled-4 and low-density lipoprotein receptor protein-5, respectively, are associated with autosomal dominant FEVR.38, 39 Functional studies have shown that NDP serves as a ligand and binds with high affinity to frizzled-4 membrane receptor, which in turn activates the canonical Wnt signaling pathway. Furthermore, the norrin–FZD4 signaling system has a central role in vascular development in the eye and ear.40
Conclusion
The identification of three families segregating recessive hearing loss due to mutations in genes present on the X-chromosome among the 1000 Pakistani families did not reveal a novel locus, an observation consistent with many similar studies in other populations.6, 14 In combination, these data suggest that most or all of the X-linked genes, which when mutated cause nonsyndromic deafness, have already been identified. Admittedly there may be an ascertainment bias, but if not, our data also suggest that there is a low prevalence of X-linked hearing impairment (0.3%; 95% confidence interval: 0.1–0.8%) in Pakistan.
Mutation nomenclature
The A nucleotide of the start codon (ATG) of GenBank POU3F4 and NDP cDNA sequences (NM_000307, NM_000266; respectively) is nucleotide +1.
References
Cohen, M. M. & Gorlin, R. J. Epidemiology Etiology and Genetic Patterns (Oxford University Press, Oxford, 1995).
Petersen, M. B., Wang, Q. & Willems, P. J. Sex-linked deafness. Clin. Genet. 73, 14–23 (2008).
de Kok, Y. J., van der Maarel, S. M., Bitner-Glindzicz, M., Huber, I., Monaco, A. P., Malcolm, S. et al. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 267, 685–688 (1995).
de Brouwer, A. P., van Bokhoven, H., Nabuurs, S. B., Arts, W. F., Christodoulou, J. & Duley, J. PRPS1 mutations: four distinct syndromes and potential treatment. Am. J. Hum. Genet. 86, 506–518 (2010).
Mathis, J. M., Simmons, D. M., He, X., Swanson, L. W. & Rosenfeld, M. G. Brain 4: a novel mammalian POU domain transcription factor exhibiting restricted brain-specific expression. EMBO J. 11, 2551–2561 (1992).
Li, J., Cheng, J., Lu, Y., Chen, A., Sun, Y., Kang, D. et al. Identification of a novel mutation in POU3F4 for prenatal diagnosis in a Chinese family with X-linked nonsyndromic hearing loss. J. Genet. Genomics 37, 787–793 (2010).
Bleeker-Wagemakers, L. M., Friedrich, U., Gal, A., Wienker, T. F., Warburg, M. & Ropers, H. H. Close linkage between Norrie disease, a cloned DNA sequence from the proximal short arm, and the centromere of the X chromosome. Hum. Genet. 71, 211–214 (1985).
Berger, W., Meindl, A., van de Pol, T. J., Cremers, F. P., Ropers, H. H., Doerner, C. et al. Isolation of a candidate gene for Norrie disease by positional cloning. Nat. Genet. 2, 84 (1992).
Meindl, A., Berger, W., Meitinger, T., van de Pol, D., Achatz, H., Dorner, C. et al. Norrie disease is caused by mutations in an extracellular protein resembling C-terminal globular domain of mucins. Nat. Genet. 2, 139–143 (1992).
Chen, Z. Y., Battinelli, E. M., Fielder, A., Bundey, S., Sims, K., Breakefield, X. O. et al. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat. Genet. 5, 180–183 (1993).
Ahmed, Z. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z., Khan, S., Griffith, A. J. et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am. J. Hum. Genet. 69, 25–34 (2001).
Wang, Q. J., Li, Q. Z., Rao, S. Q., Zhao, Y. L., Yuan, H., Yang, W. Y. et al. A novel mutation of POU3F4 causes congenital profound sensorineural hearing loss in a large Chinese family. Laryngoscope 116, 944–950 (2006).
Vore, A. P., Chang, E. H., Hoppe, J. E., Butler, M. G., Forrester, S., Schneider, M. C. et al. Deletion of and novel missense mutation in POU3F4 in 2 families segregating X-linked nonsyndromic deafness. Arch. Otolaryngol. Head Neck Surg. 131, 1057–1063 (2005).
Lee, H. K., Lee, S. H., Lee, K. Y., Lim, E. J., Choi, S. Y., Park, R. K. et al. Novel POU3F4 mutations and clinical features of DFN3 patients with cochlear implants. Clin. Genet. 75, 572–575 (2009).
de Kok, Y. J., Merkx, G. F., van der Maarel, S. M., Huber, I., Malcolm, S., Ropers, H. H. et al. A duplication/paracentric inversion associated with familial X-linked deafness (DFN3) suggests the presence of a regulatory element more than 400 kb upstream of the POU3F4 gene. Hum. Mol. Genet. 4, 2145–2150 (1995).
Bitner-Glindzicz, M., Turnpenny, P., Hoglund, P., Kaariainen, H., Sankila, E. M., van der Maarel, S. M. et al. Further mutations in Brain 4 (POU3F4) clarify the phenotype in the X-linked deafness DFN3. Hum. Mol. Genet. 4, 1467–1469 (1995).
Collin, R. W., Chellappa, R., Pauw, R. J., Vriend, G., Oostrik, J., van Drunen, W. et al. Missense mutations in POU4F3 cause autosomal dominant hearing impairment DFNA15 and affect subcellular localization and DNA binding. Hum. Mutat. 29, 545–554 (2008).
Phelps, P. D., Reardon, W., Pembrey, M., Bellman, S. & Luxom, L. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiology 33, 326–330 (1991).
Song, M. H., Lee, H. K., Choi, J. Y., Kim, S., Bok, J. & Kim, U. K. Clinical evaluation of DFN3 patients with deletions in the POU3F4 locus and detection of carrier female using MLPA. Clin. Genet. 78, 524–532 (2010).
Marlin, S., Moizard, M. P., David, A., Chaissang, N., Raynaud, M., Jonard, L. et al. Phenotype and genotype in females with POU3F4 mutations. Clin. Genet. 76, 558–563 (2009).
Samadi, D. S., Saunders, J. C. & Crenshaw, E. B. III Mutation of the POU-domain gene Brn4/Pou3f4 affects middle-ear sound conduction in the mouse. Hear. Res. 199, 11–21 (2005).
Cremers, C. W., Hombergen, G. C. & Wentges, R. T. Perilymphatic gusher and stapes surgery. A predictable complication? Clin. Otolaryngol. Allied Sci. 8, 235–240 (1983).
Staropoli, J. F., Xin, W. & Sims, K. B. Co-segregation of Norrie disease and idiopathic pulmonary hypertension in a family with a microdeletion of the NDP region at Xp11.3–p11.4. J. Med. Genet. 47, 786–790 (2010).
Gal, A., Wieringa, B., Smeets, D. F., Bleeker-Wagemakers, L. & Ropers, H. H. Submicroscopic interstitial deletion of the X chromosome explains a complex genetic syndrome dominated by Norrie disease. Cytogenet. Cell Genet. 42, 219–224 (1986).
Sims, K. B., Ozelius, L., Corey, T., Rinehart, W. B., Liberfarb, R., Haines, J. et al. Norrie disease gene is distinct from the monoamine oxidase genes. Am. J. Hum. Genet. 45, 424–434 (1989).
Zhu, D. P., Antonarakis, S. E., Schmeckpeper, B. J., Diergaarde, P. J., Greb, A. E. & Maumenee, I. H. Microdeletion in the X-chromosome and prenatal diagnosis in a family with Norrie disease. Am. J. Med. Genet. 33, 485–488 (1989).
Collins, F. A., Murphy, D. L., Reiss, A. L., Sims, K. B., Lewis, J. G., Freund, L. et al. Clinical, biochemical, and neuropsychiatric evaluation of a patient with a contiguous gene syndrome due to a microdeletion Xp11.3 including the Norrie disease locus and monoamine oxidase (MAOA and MAOB) genes. Am. J. Med. Genet. 42, 127–134 (1992).
Rodriguez-Revenga, L., Madrigal, I., Alkhalidi, L. S., Armengol, L., Gonzalez, E., Badenas, C. et al. Contiguous deletion of the NDP, MAOA, MAOB, and EFHC2 genes in a patient with Norrie disease, severe psychomotor retardation and myoclonic epilepsy. Am. J. Med. Genet. A 143A, 916–920 (2007).
Chen, Z. Y., Hendriks, R. W., Jobling, M. A., Powell, J. F., Breakefield, X. O., Sims, K. B. et al. Isolation and characterization of a candidate gene for Norrie disease. Nat. Genet. 1, 204–208 (1992).
Royer, G., Hanein, S., Raclin, V., Gigarel, N., Rozet, J. M., Munnich, A. et al. NDP gene mutations in 14 French families with Norrie disease. Hum. Mutat. 22, 499 (2003).
Schuback, D. E., Chen, Z. Y., Craig, I. W., Breakefield, X. O. & Sims, K. B. Mutations in the Norrie disease gene. Hum. Mutat. 5, 285–292 (1995).
Wu, W. C., Drenser, K., Trese, M., Capone, A. Jr & Dailey, W. Retinal phenotype-genotype correlation of pediatric patients expressing mutations in the Norrie disease gene. Arch. Ophthalmol. 125, 225–230 (2007).
Meire, F. M., Lafaut, B. A., Speleman, F. & Hanssens, M. Isolated Norrie disease in a female caused by a balanced translocation t(X,6). Ophthalmic. Genet. 19, 203–207 (1998).
Sims, K. B., Irvine, A. R. & Good, W. V. Norrie disease in a family with a manifesting female carrier. Arch. Ophthalmol. 115, 517–519 (1997).
Yamada, K., Limprasert, P., Ratanasukon, M., Tengtrisorn, S., Yingchareonpukdee, J., Vasiknanonte, P. et al. Two Thai families with Norrie disease (ND): association of two novel missense mutations with severe ND phenotype, seizures, and a manifesting carrier. Am. J. Med. Genet. 100, 52–55 (2001).
Criswick, V. G. & Schepens, C. L. Familial exudative vitreoretinopathy. Am. J. Ophthalmol. 68, 578–594 (1969).
de Crecchio, G., Simonelli, F., Nunziata, G., Mazzeo, S., Greco, G. M., Rinaldi, E. et al. Autosomal recessive familial exudative vitreoretinopathy: evidence for genetic heterogeneity. Clin. Genet. 54, 315–320 (1998).
Robitaille, J., MacDonald, M. L., Kaykas, A., Sheldahl, L. C., Zeisler, J., Dube, M. P. et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 32, 326–330 (2002).
Toomes, C., Bottomley, H. M., Jackson, R. M., Towns, K. V., Scott, S., Mackey, D. A. et al. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am. J. Hum. Genet. 74, 721–730 (2004).
Ye, X., Wang, Y. & Nathans, J. The Norrin/Frizzled4 signaling pathway in retinal vascular development and disease. Trends Mol. Med. 16, 417–425 (2010).
Huebner, A. K., Gandia, M., Frommolt, P., Maak, A., Wicklein, E. M., Thiele, H. et al. Nonsense mutations in SMPX, encoding a protein responsive to physical force, result in X-chromosomal hearing loss. Am. J. Hum. Genet. 88, 621–627 (2011).
Schraders, M., Haas, S. A., Weegerink, N. J. D., Oostrik, J., Hu, H., Hoefsloot, L. H. et al. Next-generation sequencing identifies mutations of SMPX, which encodes the small muscle protein, X-linked, as a cause of progressive hearing impairment. Am. J. Hum. Genet 88, 628–634 (2011).
Acknowledgements
We are grateful to the families who made this research possible. This work was supported by Cincinnati Children's Hospital Research Foundation (CCHMC) intramural research funds to Sheikh Riazuddin and ZMA, the National Institute on Deafness and Other Communication Disorders (NIDCD/NIH) research Grant R00-DC009287-03, a Career Development Award from RPB to ZMA. Work in Pakistan was supported by the Higher Education Commission to Sheikh Riazuddin, Islamabad; EMRO/WHO-COMSTECH and Ministry of Science and Technology (MoST) to Sheikh Riazuddin, Islamabad; the International Center for Genetic Engineering and Biotechnology, Trieste, Italy under project CRP/PAK08-01 Contract no. 08/009 to Sheikh Riazuddin. Part of this study carried out in the USA was supported by intramural funds from the NIDCD/NIH (Z01-DC00039-14) to TBF.
Note : While this manuscript was in press, two independent studies published in the May 2011 issue of American Journal of Human Genetics,41, 42 reported mutations of SMPX as another cause of X-linked nonsyndromic hearing loss in humans.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Waryah, A., Ahmed, Z., Binder, M. et al. Molecular and clinical studies of X-linked deafness among Pakistani families. J Hum Genet 56, 534–540 (2011). https://doi.org/10.1038/jhg.2011.55
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.55
Keywords
This article is cited by
-
Molecular genetic landscape of hereditary hearing loss in Pakistan
Human Genetics (2022)
-
Clinical and molecular characterization of POU3F4 mutations in multiple DFNX2 Chinese families
BMC Medical Genetics (2018)
-
Genetic Testing of Non-familial Deaf Patients for CIB2 and GJB2 Mutations: Phenotype and Genetic Counselling
Biochemical Genetics (2017)
