
Abstract
Loeys–Dietz syndrome (LDS) is an inherited disorder that is characterized by the triad of arterial tortuosity and aneurysms, hypertelorism and a bifid uvula or cleft palate. The disease is caused by heterozygous mutations in the genes encoding transforming growth factor β receptors 1 and 2 (TGFBR1 and TGFBR2, respectively). However, studies of patients with LDS are limited in Korea. From June 2000 to December 2010, 13 patients (10 probands) diagnosed with LDS were enrolled. The multidisciplinary data of the patients were reviewed retrospectively. The frequency of each clinical manifestation in Korean patients with LDS was compared with Western populations as described in the report by Loeys et al. Twelve (92%) of the 13 LDS patients had arterial tortuosity, 9 (69%) patients had hypertelorism and 11 (85%) patients had bifid uvula or cleft palate. Mutations in either TGFBR1 or TGFBR2 were detected in nine probands (90%). Of the mutations, five novel mutations were detected; three in TGFBR2 and two in TGFBR1. Blue sclera and atrial septal defect were not observed in the Korean patients, and the frequency of blue sclera was significantly lower in our Korean population than previously-described Western population (0 vs 40%; P=0.005). Despite the restricted number of patients in our study, we identified five novel mutations in the TGFBR1 and TGFBR2 genes and, except for blue sclera, no differences in phenotype are apparent between Korean patients and Western patients.
Similar content being viewed by others


Introduction
Loeys–Dietz syndrome (LDS), a genetic disorder affecting the connective tissue, is caused by mutations in the genes encoding transforming growth factor β receptors 1 and 2 (TGFBR1 and TGFBR2, respectively).1 LDS is characterized by the triad of hypertelorism, a bifid uvula or cleft palate and generalized arterial tortuosity with widespread vascular aneurysm and dissection.2 The diagnosis of LDS is made according to clinical presentations in the proband and family members, and molecular genetic testing of the TGFBR1 and TGFBR2 genes.3 Early and accurate diagnosis is essential to patients with LDS because they are at increased risk for aortic dissection or rupture at an early age. To date, most studies related to LDS are based on data obtained from the European and American populations. There are few data regarding the clinical and genetic characteristics of the Korean patients with LDS, leaving doubt as to whether or not there are racial differences in the physical presentation of LDS between the Oriental and Western populations.
Therefore, we investigated the clinical and genetic characteristics in the Korean patients with LDS, and compared clinical manifestations between Korean patients and Western populations as reported by Loeys et al.2
Materials and methods
Subjects
We reviewed 22 consecutive patients who were suspicious of having LDS from June 2000 to December 2010. All had undergone genetic analysis of the TGFBR1 and TGFBR2. Of them, we examined 13 Korean patients who were diagnosed with LDS. The diagnoses of these patients were made based on clinical presentation, physical examinations, radiological findings and genetic tests. Information about deceased family members, as well as other family members who were suspected of being affected, was obtained from the probands. Clinical data were obtained from clinical database and medical records. This study received Institutional Review Board approval, and informed consent was waived for this retrospective study.
DNA sequence analysis
Genomic DNA was extracted from peripheral blood leukocytes with the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA), following the manufacturer's instructions. The coding exons and the flanking introns of the TGFBR1 and TGFBR2 on chromosome 9q33-q34 and 3p22, respectively, were amplified using primers designed by the authors (available upon request). Direct sequencing was performed with the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA, USA) on an ABI 3100 Genetic Analyzer (Applied Biosystems).
Definitions
The methods used for assessing clinical and radiological examinations and cutoff limits are described below. To evaluate aortic root aneurysm, we measured the aortic diameter at the level of the sinuses of Valsalva from at least two images of transthoracic echocardiography. The diameter was corrected for age and body surface area, and interpreted as a Z-score.4 If transthoracic echocardiographic evaluations were not available, we measured at the level of the sinuses of Valsalva on multi-detector computed tomography angiography. Aortic root aneurysm was defined as an aortic diameter at the sinus of Valsalva ⩾2 Z-score. The diagnosis of ectopia lentis was based on slit-lamp examination after maximal dilatation of the pupil. Scoliosis was evaluated using coronal reformatted multi-detector computed tomography images. The Lippman–Cobb method was used to measure the degree of scoliotic curvature with a Cobb angle >20° representing a positive finding.
Statistical analysis
The significances of differences in percentages were evaluated using Fisher’s exact test with SPSS software (version 17.0;. SPSS Inc., Chicago, IL, USA). Descriptive statistics are reported as medians (ranges). Statistical significance was determined as a P-value <0.05.
Results
Clinical findings
Clinical features of the 13 Korean patients are summarized in Table 1. The 13 patients, six males and seven females, ranged in age from 5 to 56 years, with a median age of 24 years at last follow-up. Aortic root aneurysm or dissection was present in 12 (92%) patients. Nine patients (69%) underwent graft surgery in the ascending aorta at a median age of 17 years. Arterial tortuosity was found in 12 (92%) patients. Cleft palate or abnormal uvula was found in 11 (85%) patients. Hypertelorism, craniosynostosis, arachnodactyly and scoliosis were found in nine (69%) patients. Pectus deformity and joint laxity were found in eight patients, patent ductus arteriosus in four and dural ectasia in three. Blue sclera and atrial septal defect were not observed in this study. None of our patients had ectopia lentis, but three (23%) had dolichostenomelia, findings that are typical of Marfan syndrome. Of the 13 patients, 2 (patient 3 and 10-II) fulfilled the Ghent nosology and 1 (patient 3) fulfilled the revised Ghent nosology for Marfan syndrome.
Table 2 summarizes the frequency of phenotypic manifestations in the Korean and Western patients. Incidences of hypertelorism, cleft palate/bifid uvula, aortic root aneurysm/dissection, arterial tortuosity and other clinical manifestations described by Loeys et al.2 were not significantly different between the Korean and Western populations. The frequency of blue sclera was significantly lower in the Korean population than in the Western population (0 vs 40%; P=0.005). Ectopia lentis was not observed either in the Korean or Western populations.
Genetic analysis
The nature and location of each detected mutation is shown in Figure 1. We identified heterogenous mutations in the TGFBR1 or TGFBR2 genes in 9 out of 10 probands. TGFBR2 mutations were found in six probands and TGFBR1 mutations were found in three probands. The mutations in probands 2–9 occurred de novo, whereas mutation p.Arg495X segregated with disease in proband 1. Pedigrees of the family 1 and 10 are shown in Figure 2. Four mutations have previously been described in the literature including three in TGFBR2 (p.Arg495X,2 p.Ser449Phe,5 p.Trp504X6) and one in TGBFR1 (p.Arg487Gln7). Four of the five novel mutations identified in this study were missense mutations in the serine–threonine kinase domain mutations including p.Asp266Tyr (c.796G>T) and p.Thr375Arg (c.1124C>T) in the TGFBR1, and p.His378Arg (c.1130A>G) and p.Cys514Arg (c.1540T>C) in the TGFBR2. The remaining mutation was a complex deletion-insertion mutation in the TGFBR2 gene with preservation of the open reading frame (c.915_916delCCinsTCATG; p.Gln306delinsHisGlu). Additionally, we performed direct sequencing analysis of the fibrillin-1 gene (FBN1) in eight patients and detected no mutations.

Mutations in the TGFBR2 and the TGFBR1 genes. Novel mutations are indicated in red type. The different exons are numbered and intervening sequences are shown as black lines. The colored boxes represent the exons encoding the extracellular domain of the receptor (yellow), the transmembrane domain (green) and the serine–threonine kinase domain (blue). TGFBR2 mutations were found in seven patients (six probands), and TGFBR1 mutations were found in three patients (three probands). A full color version of this figure is available at the Journal of Human Genetics journal online.

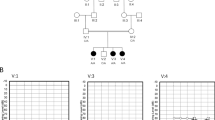
Pedigrees of the family of proband 1 and 10. Squares indicate male family members, circles indicate female family members, and symbols with slashes indicate deceased family members. Open symbols indicate unaffected family members, filled symbols indicate probands, and shaded symbols indicate family members who are or were probably affected. (a) Pedigree of the family of proband 1. Individual 1 suffered from acute aortic dissection type A at the age of 48 and died a sudden death at the age of 50. Individual 2 and 3 died a sudden death at the age of 4 and the month of 7, respectively. (b) Pedigree of the family of proband 10. Individual 1 died of non-cardiovascular disease at the age of 34, individual 2 suffered from acute aortic dissection type A at the age of 42 and individual 3 died a sudden death at the age of 22.
Discussion
In the present study, we describe 13 patients from 10 families with LDS characterized by hypertelorism, bifida uvula or cleft palate, and generalized arterial tortuosity with ascending aortic aneurysm and dissection. All 13 patients showed the typical manifestations of LDS with variable individual expressions of the clinical features. Incidences of clinical manifestations, except for blue sclera, did not show significant differences between the Korean and Western populations. Furthermore, we found three novel mutations in the TGFBR2 gene and two novel mutations in the TGFBR1 gene.
Clinical findings
Accurate diagnosis of LDS has important medical and personal implications for patients, yet diagnostic criteria have not been well established. In current practice, the diagnosis of LDS is based on characteristic clinical findings in the proband and family members, and confirmation by molecular genetic testing of the TGFBR1 and TGFR2 genes.3 However, to date, characteristic clinical findings are based on data obtained from Europeans and Americans, and data supporting racial differences in patients with LDS from other populations have not been documented. Although the relatively small size of our study population have influenced our results, our data indicate that the frequencies of clinical manifestations in our patients and in patients described by Loeys et al. are similar, except for blue sclera. These similarities may reflect the fact that characters varying across populations, such as height, arm span and body weight, are not considered in the diagnosis for LDS. Therefore, our observations suggest that clinical findings derived from the analyses of Caucasians are sufficient for diagnosing the Korean patients.
In previous studies, ∼75% of affected individuals had LDS type I with craniofacial manifestations (bifida uvula/cleft palate, craniosynostosis and hypertelorism) and ∼25% had LDS type II with cutaneous manifestations (velvety and translucent skin, easy bruising and atrophic scar), whereas all of our patients had features typical of LDS type I.3, 8 To our knowledge, in previous studies of Oriental populations, patients with LDS type II were not identified.9, 10, 11, 12, 13 Accordingly, a large-scale clinical study is needed to identify whether differences in the frequencies of LDS type I and II can be attributed to racial differences.
LDS has many similarities to Marfan syndrome with regard to cardiovascular disorders or skeletal manifestations.14, 15 In fact, two patients in our study fulfilled the Ghent criteria for Marfan syndrome and one patient fulfilled the revised Ghent criteria. One patient (patient 3) was confirmed by having no mutations in the fibrillin-1 gene and mutation of TGFBR2. The other patient (patient 10-II) was suspected LDS as he did not reveal TGFBR1 or TGFBR2 mutation despite conventional clinical findings and a positive family history.
Genetic analysis
Due to difficulties in the clinical approach to patients with various vascular involvements, with or without aortic root dilatation and LDS cardinal features, genetic analysis is essential to clarify the diagnosis and to assess the management and prognosis of the patient.11, 16 In our study, although 10 patients with the TGFBR mutations had phenotypic variability, genetic analysis can be used to differentiate LDS from Marfan syndrome or from familial thoracic aortic aneurysm and dissection. Prior studies have suggested that some TGFBR2 mutations are present in families whose members have not only LDS (p.Arg495X),2 but also classic Marfan syndrome (p.Ser449Phe),5 or incomplete Marfan syndrome (p.Trp504X)6 and TGFBR1 mutations are present in patients with familial thoracic aortic aneurysm and dissection (p.Arg487Gln).7 However, our patients with the p.Arg495X, p.Ser449Phe, p.Trp504X and p.Arg487Gln had features typical of LDS type I. These findings suggest that patients who carried the same mutation as the affected proband could still exhibit phenotypic variability. In addition, the novel mutations that were found in this study further expand the spectrum of known TGFBR1 and TGFBR2 mutations in LDS patients.
In agreement with previous reports, the mutations were located on the intracellular kinase domain of the receptor in exon 4–7 of the TGFBR2 gene and exon 4, 6 and 9 of the TGFBR1 gene.1, 5, 7, 10, 12 Mutations found in the TGFBR gene associated with LDS are essentially missense and only rare instances of nonsense or deletions have been identified.2, 6 In our study, two probands had a mutation leading to a premature termination codon, whereas seven probands had a mutation leading to an altered protein. Interestingly, family 1 and patient 5, both with a nonsense mutation of TGFBR2, had few systemic manifestations and underwent their first cardiovascular surgery at a late age. Patient 1-II suffered from acute aortic dissection type B at the age of 33, and patient 5 suffered from acute aortic dissection type A at the age of 45. Our study and previous studies show that patients with nonsense mutations of either the TGFBR1 or TGFBR2 genes are generally older than patients with missense mutations, with the exception of one 7-year-old patient (Table 3). On the other hand, the seven patients with missense mutations of the TGFBR1 or TGFBR2 genes who underwent graft surgery in the ascending aorta due to acute root aneurysm or dissection ranged in age from 4 to 22 years, with a median age of 11. Accordingly, further studies of a large sample are needed to understand the relationship between the mutation type and the phenotype, and to identify whether nonsense mutation of TGFBR may have milder systemic features and later-onset cardiovascular event than missense mutation of TGFBR.
Patients in the family of proband 10 did not show mutations in TGFBR despite having clinical features typical of LDS. It is reported that 5% of LDS patients are negative in the genetic study of TGFBR1 or TGFBR2 mutations.3, 17 Theoretically, sequence analysis of these two genes can only detect point mutations so that a large deletion or duplication, deep intronic mutation, and promoter site mutations may be possible to miss a diagnosis for LDS. However, as of yet, family 10 should be regarded as a suspected LDS patient as there has been no report that a large deletion or duplication, deep intronic mutation or promoter site mutation was found in LDS case.
Our study has several limitations. First, our study sample is unlikely to represent an unbiased fraction of individuals from the general population, as many of the enrolled patients were referred to our hospital to undergo aortic surgery. Second, the relatively small size of our study population may have influenced our results. Therefore, further studies with larger numbers of patients are needed to verify clinical and genetic features of LDS in the Korean patients. Such studies will help clinicians to better understand this serious, but overlooked, disorder and to develop more appropriate therapeutic strategies.
In conclusion, although the number of the patients was limited, we identified five novel mutations in the TGFBR1 and TGFBR2 genes in 13 patients affected with LDS. We suggested that no differences in phenotype are apparent between Korean patients and Western patients, except for the manifestation of blue sclera.
References
Loeys, B. L., Chen, J., Neptune, E. R., Judge, D. P., Podowski, M., Holm, T. et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 37, 275–281 (2005).
Loeys, B. L., Schwarze, U., Holm, T., Callewaert, B. L., Thomas, G. H., Pannu, H. et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 355, 788–798 (2006).
Van Hemelrijk, C., Renard, M. & Loeys, B. The Loeys-Dietz syndrome: an update for the clinician. Curr. Opin. Cardiol. 25, 546–551 (2010).
Roman, M. J., Devereux, R. B., Kramer-Fox, R. & O’Loughlin, J. Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am. J. Cardiol. 64, 507–512 (1989).
Mizuguchi, T., Collod-Beroud, G., Akiyama, T., Abifadel, M., Harada, N., Morisaki, T. et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet. 36, 855–860 (2004).
Stheneur, C., Collod-Beroud, G., Faivre, L., Gouya, L., Sultan, G., Le Parc, J. M. et al. Identification of 23 TGFBR2 and 6 TGFBR1 gene mutations and genotype-phenotype investigations in 457 patients with Marfan syndrome type I and II, Loeys-Dietz syndrome and related disorders. Hum. Mutat. 29, E284–295 (2008).
Matyas, G., Arnold, E., Carrel, T., Baumgartner, D., Boileau, C., Berger, W. et al. Identification and in silico analyses of novel TGFBR1 and TGFBR2 mutations in Marfan syndrome-related disorders. Hum. Mutat. 27, 760–769 (2006).
Yang, J. H., Lee, S. T., Kim, J. A., Kim, S. H., Jang, S. Y., Ki, C. S. et al. Genetic analysis of three Korean patients with clinical features of Ehlers-Danlos syndrome type IV. J Korean Med. Sci. 22, 698–705 (2007).
Togashi, Y., Sakoda, H., Nishimura, A., Matsumoto, N., Hiraoka, H. & Matsuzawa, Y. A Japanese family of typical Loeys-Dietz syndrome with a TGFBR2 mutation. Intern. Med. 46, 1995–2000 (2007).
Akutsu, K., Morisaki, H., Takeshita, S., Sakamoto, S., Tamori, Y., Yoshimuta, T. et al. Phenotypic heterogeneity of Marfan-like connective tissue disorders associated with mutations in the transforming growth factor-beta receptor genes. Circ. J. 71, 1305–1309 (2007).
Sakai, H., Visser, R., Ikegawa, S., Ito, E., Numabe, H., Watanabe, Y. et al. Comprehensive genetic analysis of relevant four genes in 49 patients with Marfan syndrome or Marfan-related phenotypes. Am. J. Med. Genet. A 140, 1719–1725 (2006).
Ki, C. S., Jin, D. K., Chang, S. H., Kim, J. E., Kim, J. W., Park, B. K. et al. Identification of a novel TGFBR2 gene mutation in a Korean patient with Loeys-Dietz aortic aneurysm syndrome; no mutation in TGFBR2 gene in 30 patients with classic Marfan's syndrome. Clin. Genet. 68, 561–563 (2005).
Muramatsu, Y., Kosho, T., Magota, M., Yokotsuka, T., Ito, M., Yasuda, A. et al. Progressive aortic root and pulmonary artery aneurysms in a neonate with Loeys-Dietz syndrome type 1B. Am. J. Med. Genet. A 152A, 417–421 (2010).
Erkula, G., Sponseller, P. D., Paulsen, L. C., Oswald, G. L., Loeys, B. L. & Dietz, H. C. Musculoskeletal findings of Loeys-Dietz syndrome. J. Bone Joint Surg. Am. 92, 1876–1883 (2010).
Soylen, B., Singh, K. K., Abuzainin, A., Rommel, K., Becker, H., Arslan-Kirchner, M. et al. Prevalence of dural ectasia in 63 gene-mutation-positive patients with features of Marfan syndrome type 1 and Loeys-Dietz syndrome and report of 22 novel FBN1 mutations. Clin. Genet. 75, 265–270 (2009).
Drera, B., Ritelli, M., Zoppi, N., Wischmeijer, A., Gnoli, M., Fattori, R. et al. Loeys-Dietz syndrome type I and type II: clinical findings and novel mutations in two Italian patients. Orphanet. J. Rare Dis. 4, 24 (2009).
Pagon, R. A., Bird, T. D., Dolan, C. R. & Stephens, K. (eds) GeneReviews (Internet), University of Washington: Seattle (WA), 1993.
Acknowledgements
We thank the patients for their participation in this study and members of the Cardiac and Vascular Center, Department of Laboratory Medicine and Genetics, Department of Thoracic and Cardiovascular Surgery, Department of Orthopedic Surgery, Department of Pediatrics and Department of Ophthalmology.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Yang, J., Ki, CS., Han, H. et al. Clinical features and genetic analysis of Korean patients with Loeys–Dietz syndrome. J Hum Genet 57, 52–56 (2012). https://doi.org/10.1038/jhg.2011.130
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.130
Keywords
This article is cited by
-
Loeys–Dietz syndrome in a Southeast Asian Hospital: a case series
European Journal of Pediatrics (2014)
