
Abstract
Mitochondrial DNA (mtDNA) depletion syndrome encompasses a heterogeneous group of disorders characterized by a reduction in the mtDNA copy number. We identified two patients with clinical presentations consistent with mtDNA depletion syndrome (MDS), who were subsequently found to have apparently homozygous point mutations in TYMP and DGUOK, two of the nine nuclear genes commonly associated with these disorders. Further sequence analyses of parents indicated that in each case only one parent; the mother of the first and the father of the second, was a heterozygous carrier of the mutation identified in the affected child. The presence of underlying deletions was ruled out by use of a custom target array comparative genomic hybridization (CGH) platform. A high-density single-nucleotide polymorphism (SNP) array analysis revealed that the first patient had a region of copy-neutral absence of heterozygosity (AOH) consistent with segmental isodisomy for an 11.3 Mb region at the long-arm terminus of chromosome 22 (including the TYMP gene), and the second patient had results consistent with complete isodisomy of chromosome 2 (where the DGUOK gene is located). The combined sequencing, array CGH and SNP array approaches have demonstrated the first cases of MDS due to uniparental isodisomy. This diagnostic scenario also demonstrates the necessity of comprehensive examination of the underlying molecular defects of an apparently homozygous mutation in order to provide patients and their families with the most accurate molecular diagnosis and genetic counseling.
Similar content being viewed by others


Introduction
Mitochondrial disorders may be caused by mutations in mitochondrial DNA (mtDNA) or in nuclear genes having roles in mitochondrial biogenesis, structure and function. To date, nine nuclear genes, namely C10orf2, DGUOK, MPV17, POLG, RRM2B, SUCLA2, SUCLG1, TK2 and TYMP, are known to cause mtDNA depletion syndrome (MDS), which represents a specific subset of mitochondrial disorders characterized by reduced mtDNA copy number.1, 2 The protein products of these genes are involved in the maintenance of mtDNA integrity, including mtDNA replication and the supply of nucleotides by salvage pathways. Three different clinical disorders have been described based upon the main organ systems affected: myopathic, encephalomyopathic and hepatocerebral. In general, TK2 and RRM2B are associated with the myopathic form; SUCLG1 and SUCLA2 with the encephalomyopathic form; and C10orf2, DGUOK, MPV17 and POLG with the hepatocerebral form.1 However, there is considerable variability, such that SUCLA2 mutations may have a predominantly myopathic presentation, whereas POLG mutations, in addition to Alpers syndrome, may cause more varied clinical phenotypes, including myopathy, progressive external ophthalmoplegia (PEO) and PEO-plus syndromes, or mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS).3, 4 Mutations in TYMP cause a specific syndrome, mitochondrial neurogastrointestinal encephalopathy (MNGIE), which also has mtDNA depletion as a feature.5 C10orf2 and POLG can cause mitochondrial disease via autosomal dominant mutations leading to multiple mtDNA deletions, but MDS is generally inherited in an autosomal recessive manner for all nine genes.2, 6
Comprehensive genetic testing for mitochondrial disorders is becoming broadly available, but these diseases pose unique diagnostic challenges. Although generally characterized by neuromuscular involvement, clinical heterogeneity is a complicating factor, with patients often exhibiting a multitude of apparently unrelated multisystem symptoms that can be non-specific and/or episodic. The genetic interactions that underlie these clinical manifestations are also very complex. Mutations in mtDNA itself can be sporadic or inherited and can exhibit heteroplasmy; the coexistence of mutant and wild-type mtDNA in varying percentages in different tissues and over time. Mutations in nuclear genes regulating mtDNA biosynthesis are inherited in standard Mendelian patterns, mostly autosomal recessive or dominant, but can also have secondary effects on mtDNA integrity itself, such as point mutations, multiple deletions and depletion.7, 8 Precise molecular diagnosis is thus instrumental in directing proper patient management and genetic counseling. Published testing algorithms can aid in focusing molecular testing by distinguishing mitochondrial disorders from other metabolic disturbances as well as from each other.9, 10
Once mutations are identified in an individual affected with a recessive disorder, it becomes even more pertinent to verify a chromosomal trans configuration (the phase) by parental testing. As mutations in multiple different genes may cause similar phenotypes, and heterozygous individuals may under certain circumstances be symptomatic, it is crucial that the genotype of a patient with a mitochondrial disorder be unambiguously established. This principle is most clearly illustrated in cases of apparently homozygous nuclear gene mutations, which may be explained by a variety of genetic and non-genetic mechanisms, including a failure to PCR amplify one allele and non-paternity. We present two such examples of patients with apparently homozygous mutations, where, based upon extensive familial follow-up testing, it emerged that non-Mendelian inheritance had occurred. These affected individuals represent the first reported instances of MDS due to uniparental isodisomy for the chromosomal regions containing the TYMP and DGUOK genes.
Materials and methods
Specimens from the two families described here were sent to the Medical Genetics Laboratories at Baylor College of Medicine for diagnostic genetic testing for their respective conditions between 2006 and 2009. Pertinent information from their available medical records was obtained from the Departments of Genetics at Kaiser Permanente Sacramento and San Jose, and from the Department of Neurology at Columbia University Medical Center. This study was approved by the Institutional Review Board of Baylor College of Medicine.
Total genomic DNA for testing of patient 1 and both sets of parents was isolated from blood, for patient 2 from liver, and for the sibling of patient 2 from amniocytes obtained prenatally. Archived patient samples were used, and no additional tissue sources were available at the time of analysis.
Sequence analysis was performed using PCR primers designed to amplify all coding exons and ∼50 bp of the flanking-intron regions of the DGUOK and TYMP genes. PCR fragments were analyzed by direct sequencing using ABI3730XL and BigDye v3.1 Terminators (Applied Biosystems, Foster City, CA, USA). Sequences were compared with the published NCBI sequence (GenBank accession numbers: NM_080916.1 for DGUOK and NC_000022.9 for TYMP).
An oligonucleotide-based custom array using the Agilent 4 × 44 000 microarray platform (Agilent Technologies, Santa Clara, CA, USA) was used for the detection of intragenic deletions. The array contains densely covered probes for 192 targeted genes for mitochondrial and metabolic disorders. The array also contains 6000 low-resolution backbone probes and 6400 oligonucleotide probes covering the 16.6 kb of the entire mitochondrial genome in both forward and reverse directions. This array is referred to as the MitoMet array. The array-comparative genomic hybridization (aCGH) experiment and analysis of results were performed as previously published.11, 12, 13, 14
Real-time quantitative PCR was used to measure the mtDNA content as previously described.15
The Human610-Quad single-nucleotide polymorphism (SNP) array was used as per the manufacturer's protocol (Illumina Inc., San Diego, CA, USA). Genotyping data was analyzed for copy number variations and copy-neutral absence of heterozygosity (AOH) with GenomeStudio software using CNV partition 2.3.4 algorithm (Illumina Inc.). Uniparental disomy (UPD) of chromosome 2 testing was clinically performed by the Mayo Medical Laboratories.
The Identifiler Kit (Applied Biosystems), comprised of 15 independent short tandem repeat loci and Amelogenin, was used to address the identity of the biological parents.
Results
TYMP clinical report
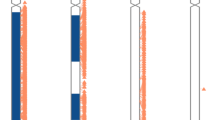
Patient 1 was born to a non-consanguineous couple of Northern European descent and was healthy in early childhood, but his parents retrospectively reported that from age 12 years, he had a poor appetite and early satiety. Bilateral ptosis was noted in early adulthood. He has two healthy, unaffected sisters. At age 22 years, he developed paresthesias in his hands and feet. Examination revealed ptosis without ophthalmoparesis, normal hearing, weakness that was more pronounced in the legs than the arms, stocking and glove sensory loss and areflexia. A few months later, he was injured in a motorcycle accident that was attributed to his peripheral neuropathy. Nerve conduction study revealed reduced motor and sensory conduction velocities, indicative of a demyelinating peripheral neuropathy. Electromyography revealed a denervation pattern. A sural nerve biopsy was reported to have been abnormal. Over the subsequent year, the patient lost ∼30 pounds and he became emaciated, weighing 85 pounds, yet with a height of 6 feet. In addition, he became weaker such that he was virtually quadriplegic and bed-ridden. TPN was administered for one year, but was stopped due to abnormal liver function tests. The following tests were normal or negative: molecular testing for Charcot–Marie–Tooth disease, HIV, Lyme disease, heavy metals, very-long-chain fatty acids and B12. Brain magnetic resonance imaging revealed diffuse white matter changes. He suffered a variety of complications, including sinus tachycardia, chronic pain from dysesthesias, diabetes, chronic diarrhea and a variety of secondary infections, including urinary tract infections and aspiration pneumonia. He received multiple medications, including gabapentin, cyclophosphamide, immunoglobulin infusion, antibiotics and plasmapheresis. He was hospitalized multiple times and, at age 24, he was admitted to the intensive care unit for palliative care and pain management and died. Just before death, a blood sample revealed severely reduced activity of thymidine phosphorylase in a buffy coat preparation (42.1 nmol h−1 per mg protein; normal range 200–1340) and greatly elevated levels of plasma thymidine (18.2 μmol; normal <0.05), thus confirming the diagnosis of mitochondrial neurogastrointestinal encephalopathy. Molecular testing revealed an apparently homozygous missense mutation, c.457G>A (p.G153S) in the TYMP gene. Carrier testing was performed on both parents, but surprisingly only identified the pathogenic mutation in the mother and not in the father. Re-testing of the father by full gene sequencing and with a different set of PCR primers for the amplicon containing the proband's mutation did not identify any pathogenic changes. Non-paternity was addressed and ruled out using a standard set of widely used unlinked short tandem repeat markers. The c.457G>A (p.G153S) genotypes for this family are summarized in Figure 1a.

(a) Family pedigree showing the inheritance pattern of the c.457G>A TYMP mutation. The deceased proband, patient 1, is homozygous for this change, the mother is heterozygous and the father does not carry the mutation. The two older unaffected sisters were not tested. (b) Family pedigree showing the inheritance pattern of the c.533G>A DGUOK mutation. The deceased proband, patient 2, is homozygous, the father is a heterozygous carrier, and the mother and younger sister do not carry the mutation. This sibling was tested as part of prenatal diagnosis. The older brother who was previously known to have a 48,XXXY karyotype was not tested for the familial DGUOK mutation. A full color version of this figure is available at the Journal of Human Genetics journal online.
DGUOK clinical report
Patient 2 was born following a normal pregnancy to a non-consanguineous couple from Mexico. Her birth weight was 3100 g, and length and head circumference were normal. The family history was remarkable for an older brother with a chromosome disorder, 48,XXXY. The patient was discharged soon after birth, but was re-admitted to the hospital a few days later due to hypotonia and poor feeding. Blood lactic acid during the admission was elevated at 5.4–8.8 mM (normal 0.5–2.2 mM). Blood ammonia and ALT were normal. Urine organic acids showed markedly elevated tyrosine metabolites, but urine succinylacetone was not detected. Based upon newborn screening, blood tyrosine was found to be moderately elevated. A physical examination at 12 days of age demonstrated a normal liver size and a normal neurological examination. Plasma amino acids at 13 days of age revealed an elevated tyrosine (1018 μmol l−1, normal 40–110 μmol l−1). Because of the elevated tyrosine she was treated with 200 mg a day of vitamin C. At 25 days of age, the plasma tyrosine remained mildly elevated (218 μmol l−1). The physical examination at 7 weeks of age was completely normal and she was clinically well, with weight, length, and head circumference at 10% for her age. The plasma tyrosine was normal. At 3 months of age, nystagmus and an enlarged liver that was 3 cm below the right costal margin were noted. ALT, total and conjugated bilirubin, and gamma glutamyl transpeptidase were mildly elevated. The urine succinylacetone was again normal, as were chest/abdomen X-rays, urine catecholamines, alpha-1-antitrypsin testing, plasma amino acids and a cholangiogram. Liver biopsy showed cirrhosis with nodules separated by thin fibrous septae almost entirely following a portal-based pattern of fibrosis. Bile ducts were normal in number. A cranial magnetic resonance imaging was normal. By 10 months of age, all growth parameters were below 3% for her age, and developmental delay and hypotonia were apparent. Testing for carbohydrate-deficient transferrin was normal. Urine organic acid analysis showed a markedly elevated lactic acid, with more modest elevations of dicarboxylic and hydroxydicarboxylic acids. At 12 months of age, she was admitted for liver failure. Despite undergoing liver transplantation, she died within a few weeks. A sample of her explanted liver was sent for POLG and DGUOK DNA sequence analyses, which identified an apparently homozygous c.533G>A (p.W178X) DGUOK nonsense mutation. The father was found to be a carrier and the mother was not. The parents were counseled that the recurrence risk for another child affected with DGUOK deficiency was very low. During a subsequent pregnancy, targeted prenatal testing for the DGUOK mutation and a routine karyotype, performed due to the previous other child with chromosome abnormality, were normal. The c.533G>A (p.W178X) genotypes for this family are summarized in Figure 1b.
Follow-up molecular analysis
Both the c.457G>A (p.G153S) change in TYMP and the c.533G>A (p.W178X) change in DGUOK have been previously reported as disease-causing mutations,16 which prompted further investigation of the probands and the non-carrier parents. In order to rule out deletions in the respective genes, targeted array CGH was performed for both the patients using the custom MitoMet platform. Large intragenic deletions were not detected in either TYMP or DGUOK (Figures 2a and c). The MitoMet platform also has the capacity to simultaneously measure mtDNA content, which was reduced in both patients relative to tissue-matched controls (Figures 2b and d). The mtDNA content in the liver of patient 2 was also measured by real-time quantitative PCR and was found to be decreased at about 5% of tissue-matched control mtDNA (data not shown). High-density SNP array analysis in patient 1 detected a simple block of copy-neutral AOH of ∼11.3 Mb at the long-arm terminus of chromosome 22, including the TYMP gene (Figure 3a), indicating presence of segmental isodisomy. Testing for UPD of chromosome 2 performed on patient 2 showed only paternal inheritance of informative markers (D2S162, D2S391, D2S2368, D2S2216, D2S112, D2S2330, D2S335, D2S396 and D2S338) and no inheritance of maternal alleles (data not shown). SNP array analysis subsequently revealed that the patient had copy-neutral AOH across the entire chromosome 2, where the DGUOK gene is located (Figure 3b). SNP analysis did not detect other large blocks of AOH elsewhere in the genomes of the two patients.

(a) TYMP copy number in patient 1. Oligonucleotides spanning the TYMP gene show no copy number alterations when hybridized to DNA from the patient and a normal control. (b) mtDNA content in patient 1. Oligonucleotides spanning the mtDNA molecule all show reduced copy number in blood as compared with that in a tissue-matched control. (c) DGUOK copy number in patient 2. Oligonucleotides spanning the DGUOK gene show no copy number alterations when hybridized to DNA from the patient and a normal control. (d) mtDNA content in patient 2. Oligonucleotides spanning the mtDNA molecule all show reduced copy number in the liver as compared with that in a tissue-matched control. A full color version of this figure is available at the Journal of Human Genetics journal online.

(a) Segmental AOH for an 11.3-Mb region at the long-arm terminus of chromosome 22 in patient 1. The location of the TYMP gene is indicated. (b) AOH for the entire chromosome 2 in patient 2. The location of the DGUOK gene is indicated. In both instances no other significant regions of AOH were detected elsewhere in the genome. The AOH regions, as demonstrated by the B allele frequencies, are shaded (upper panels). There are no copy number changes, as demonstrated by the log R ratios (lower panels). A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
The data presented here establish the first reported instances of MDS with homozygous mutations in the TYMP and DGUOK genes due to UPD for a region on chromosome 22 and the entire chromosome 2, respectively. These cases also highlight the importance of follow-up testing after clinical identification of an apparently homozygous mutation in a patient with a strong support of diagnosis. True homozygosity may be confirmed when both parents are heterozygous carriers, which may be more likely in cases of consanguinity or identity by descent in founder populations. Alternatively, a point mutation identified by PCR-based DNA sequencing may appear to be homozygous when the other allele contains a deletion spanning the region containing the mutation.17 A patient could thus be compound heterozygous for two deleterious mutations (one a point mutation and the other a deletion), consistent with affected status. The chance that a new mutation could have occurred in a patient with a recessive disorder who had already inherited an identical mutation from one of the parents, or that one parent has gonadal mosaicism, is highly unlikely, and not considered as part of our standard evaluation of apparently homozygous mutations. However, the possibility that one parent is not the biological parent may be appropriate to discuss with the family before proceeding with further molecular testing in cases of apparent homozygosity. Although non-paternity rates are reported to vary greatly between studied populations, it may be as high as 12%, making this a relatively common cause of observing inconsistent genotypes in a pedigree.18, 19, 20 Non-genetic reasons for the initial, seemingly discordant, sequencing results observed in these families may also include allele dropout due to preferential amplification of only one allele, or sample mix-up, which are controlled for in the strict diagnostic laboratory environment.
UPD describes the inheritance of a pair of homologous chromosomes from one parent without contribution from the other parent. Uniparental isodisomy specifically describes the inheritance of two identical copies of the same chromosome, and uniparental heterodisomy the inheritance of the two different homologous chromosomes from one parent. Either type of UPD can have a role in imprinting disorders if the chromosome or segment of chromosome involved contains genes differentially imprinted based on the parent of origin.21 Uniparental isodisomy can also cause recessive conditions if the chromosome or region of chromosome inherited as an identical copy harbors a disease-causing mutation. The first such case was described for a patient with cystic fibrosis and suspected Russell–Silver syndrome, who was found to have maternal isodisomy for chromosome 7.22 Since then, both heterodisomy and isodisomy forms of UPD have been reported for most human chromosomes, including 2 and 22. Prevailing evidence does not support the existence of imprinted loci on either of those chromosomes.23, 24 Consistent with these findings, the two patients described here have clinical findings typical of MDS without any obvious additional findings that may be attributed to the underlying isodisomy.
We utilized two clinically available array methodologies in our investigation of apparent homozygosity. The MitoMet and SNP arrays both provide copy number information for the two nuclear genes of interest. The MitoMet platform is the preferred method for ruling out the presence of a gene deletion, as the latest version of this custom targeted array contains exon-by-exon coverage of genes related to mitochondrial and metabolic disorders, including the nine nuclear genes currently known to cause mtDNA depletion. This array CGH-based method also allowed us to simultaneously measure mtDNA amount, thus confirming mtDNA depletion in the two patients.12, 14, 25, 26 In addition, the SNP array revealed that AOH is responsible for the observed homozygosity in both the affected individuals, even if it did not definitively clarify the mechanism underlying the AOH. SNP arrays have advantages and disadvantages compared with the well-established microsatellite marker genotyping traditionally used for the purpose of UPD analysis. SNP arrays can provide a more comprehensive view of the entire genome, not just the chromosome in question. This analysis allows the simultaneous assaying of a multitude of markers, which can more precisely define the boundaries of the AOH region, and is thus not limited by lack of informative markers. SNP arrays can also provide useful information even if one or both parents is/are not available for testing. However, unlike microsatellite analysis, unless SNP arrays are run on the family trio and the actual genotypes analyzed, the parent of origin for the observed UPD cannot be determined. However, the parent of origin of the specific mutation in question has been confirmed by the analysis of both parents.
Clinically, SNP array analysis is an efficient approach when consanguinity, identity by descent, or uniparental isodisomy is suspected. The patterns of AOH seen with uniparental isodisomy can be distinguished from those due to the other causes. AOH for one whole chromosome and no other AOH regions in the genome is strongly suggestive of isodisomy for that chromosome. This type of UPD is thought to arise by meiotic non-disjunction in one parent who does not contribute a given chromosome, followed by monosomy rescue via duplication of the chromosome contributed by the other parent.23 Our patient 2 most likely exemplifies this mechanism based on the observed chromosome 2 data. Recombination events can result in segmental isodisomy, characterized by AOH for part of a chromosome. Somatic recombination during mitosis can lead to segmental isodisomy and biparental inheritance for the rest of the chromosome. Recombination during meiosis coupled with non-disjunction and gamete complementation or trisomy rescue can result in mixed segmental isodisomy and heterodisomy.23 A SNP array performed on a patient alone cannot differentiate between uniparental and biparental heterodisomy inheritance. When full delineation of the type and mechanism of UPD is necessary, microsatellite typing or SNP array analyses on the family trio can provide additional information. Given that trisomy rescue, or loss of one of the three chromosomes from a trisomic conceptus, is believed to be the predominant mechanism leading to UPD for chromosome 22,27 this is the most likely cause for the observed segmental isodisomy in our patient 1. Although it remains unclear if the rest of chromosome 22 contains uniparental biparental heterodisomy, knowing that information does not affect the conclusion that segmental isodisomy is present over the region containing the homozygous TYMP gene mutation. Irrespective of the mechanisms that lead to the observed segmental isodisomy, it would be a sporadic event without appreciable recurrence risk. The recurrence risk for UPD and therefore the same recessive condition in a subsequent child in general is believed to be so low that invasive prenatal diagnosis is not generally indicated.28
The parents of patient 1 are past reproductive age, however, the parents of patient 2 did opt for prenatal diagnosis for the c.533G>A (p.W178X) mutation in their next child. Considering that the patient appeared to have uniparental isodisomy due to non-disjunction in the maternal germline, and that the couple had another child with a 48,XXXY karyotype, also a chromosome abnormality consistent with non-disjunction in the mother, it may be that there are additional genetic factors in this family predisposing to such events. In general, the risk to future children is much <25% risk quoted for recessive disorders when both the parents are heterozygous carriers. With the availability of SNP and combination SNP and oligonucleotide arrays, testing future pregnancies by such methodologies, in conjunction with targeted testing for a familial mutation, may offer a family such as this the most reassurance.
Accession codes
References
Spinazzola, A., Invernizzi, F., Carrara, F., Lamantea, E., Donati, A., Dirocco, M. et al. Clinical and molecular features of mitochondrial DNA depletion syndromes. J. Inherit. Metab. Dis. 32, 143–158 (2009).
Spinazzola, A. & Zeviani, M. Disorders from perturbations of nuclear-mitochondrial intergenomic cross-talk. J. Intern. Med. 265, 174–192 (2009).
Milone, M. & Massie, R. Polymerase gamma 1 mutations: clinical correlations. Neurologist 16, 84–91 (2010).
Deschauer, M., Tennant, S., Rokicka, A., He, L., Kraya, T., Turnbull, D. M. et al. MELAS associated with mutations in the POLG1 gene. Neurology 68, 1741–1742 (2007).
Nishino, I., Spinazzola, A. & Hirano, M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283, 689–692 (1999).
Spinazzola, A. & Zeviani, M. Disorders of nuclear-mitochondrial intergenomic signaling. Gene 354, 162–168 (2005).
Wong, L. J. & Boles, R. G. Mitochondrial DNA analysis in clinical laboratory diagnostics. Clin. Chim. Acta. 354, 1–20 (2005).
Wong, L. J. Diagnostic challenges of mitochondrial DNA disorders. Mitochondrion 7, 45–52 (2007).
Berardo, A., DiMauro, S. & Hirano, M. A diagnostic algorithm for metabolic myopathies. Curr. Neurol. Neurosci. Rep. 10, 118–126 (2010).
Wong, L. J., Scaglia, F., Graham, B. H. & Craigen, W. J. Current molecular diagnostic algorithm for mitochondrial disorders. Mol. Genet. Metab. 100, 111–117 (2010).
Chinault, A. C., Shaw, C. A., Brundage, E. K., Tang, L. Y. & Wong, L. J. Application of dual-genome oligonucleotide array-based comparative genomic hybridization to the molecular diagnosis of mitochondrial DNA deletion and depletion syndromes. Genet. Med. 11, 518–526 (2009).
Lee, N. C., Dimmock, D., Hwu, W. L., Tang, L. Y., Huang, W. C., Chinault, A. C. et al. Simultaneous detection of mitochondrial DNA depletion and single-exon deletion in the deoxyguanosine gene using array-based comparative genomic hybridisation. Arch. Dis. Child. 94, 55–58 (2009).
Shchelochkov, O. A., Li, F. Y., Geraghty, M. T., Gallagher, R. C., Van Hove, J. L., Lichter-Konecki, U et al. High-frequency detection of deletions and variable rearrangements at the ornithine transcarbamylase (OTC) locus by oligonucleotide array CGH. Mol. Genet. Metab. 96, 97–105 (2009).
Wong, L. J., Dimmock, D., Geraghty, M. T., Quan, R., Lichter-Konecki, U., Wang, J. et al. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clin. Chem. 54, 1141–1148 (2008).
Dimmock, D., Tang, L. Y., Schmitt, E. S. & Wong, L. J. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin. Chem. 56, 1119–1127 (2010).
Poulton, J., Hirano, M., Spinazzola, A., Arenas Hernandez, M., Jardel, C., Lombès, A. et al. Collated mutations in mitochondrial DNA (mtDNA) depletion syndrome (excluding the mitochondrial gamma polymerase, POLG1). Biochim. Biophys. Acta. 1792, 1109–1112 (2009).
Li, F. Y., El-Hattab, A. W., Bawle, E. V., Boles, R. G., Schmitt, E. S., Scaglia, F. et al. Molecular spectrum of SLC22A5 (OCTN2) gene mutations detected in 143 subjects evaluated for systemic carnitine deficiency. Hum. Mutat. 31, E1632–E1651 (2010).
Voracek, M., Haubner, T. & Fisher, M. L. Recent decline in nonpaternity rates: a cross-temporal meta-analysis. Psychol. Rep. 103, 799–811 (2008).
Cerda-Flores, R. M., Barton, S. A., Marty-Gonzalez, L. F., Rivas, F. & Chakraborty, R. Estimation of nonpaternity in the Mexican population of Nuevo Leon: a validation study with blood group markers. Am. J. Phys. Anthropol. 109, 281–293 (1999).
Sasse, G., Müller, H., Chakraborty, R. & Ott, J. Estimating the frequency of nonpaternity in Switzerland. Hum. Hered. 44, 337–343 (1994).
Engel, E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am. J. Med. Genet. 6, 137–143 (1980).
Spence, J. E., Perciaccante, R. G., Greig, G. M., Willard, H. F., Ledbetter, D. H., Hejtmancik, J. F. et al. Uniparental disomy as a mechanism for human genetic disease. Am. J. Hum. Genet. 42, 217–226 (1988).
Robinson, W. P. Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays 22, 452–459 (2000).
Kotzot, D. & Utermann, G. Uniparental disomy (UPD) other than 15: phenotypes and bibliography updated. Am. J. Med. Gene. A 136, 287–305 (2005).
Zhang, S., Li, F. Y., Bass, H. N., Pursley, A., Schmitt, E. S., Brown, B. L. et al. Application of oligonucleotide array CGH to the simultaneous detection of a deletion in the nuclear TK2 gene and mtDNA depletion. Mol. Genet. Metab. 99, 53–57 (2010).
Compton, A. G., Troedson, C., Wilson, M., Procopis, P. G., Li, F. Y., Brundage, E. K. et al. Application of oligonucleotide array CGH in the detection of a large intragenic deletion in POLG associated with Alpers syndrome. Mitochondrion 11, 104–107 (2011).
Robinson, W. P., Barrett, I. J., Bernard, L., Telenius, A., Bernasconi, F., Wilson, R. D. et al. Meiotic origin of trisomy in confined placental mosaicism is correlated with presence of fetal uniparental disomy, high levels of trisomy in trophoblast, and increased risk of fetal intrauterine growth restriction. Am. J. Hum. Genet. 60, 917–927 (1997).
Kotzot, D. Complex and segmental uniparental disomy updated. J. Med. Genet. 45, 545–556 (2008).
Acknowledgements
We would like to thank the families who made this publication possible. Special thanks to Dr Eric S Schmitt and Dr Christine M Eng for their support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Douglas, G., Wiszniewska, J., Lipson, M. et al. Detection of uniparental isodisomy in autosomal recessive mitochondrial DNA depletion syndrome by high-density SNP array analysis. J Hum Genet 56, 834–839 (2011). https://doi.org/10.1038/jhg.2011.112
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.112
